Medicinsk ekspert af artiklen

Nye publikationer
hamartom
Sidst revideret: 29.06.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
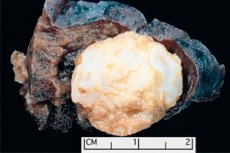
Tumorlignende formationer lokaliseret i et hvilket som helst anatomisk område som følge af unormal vækst af godartet væv defineres i medicin som hamartoma (fra græsk hamartia - fejl, defekt). [ 1 ]
Epidemiologi
Statistisk set tegner hamartomer sig for 1,2% af alle godartede neoplasmer. Prævalensen af pulmonale hamartomer anslås at være cirka 0,25% af den generelle befolkning og tegner sig for op til 8% af alle pulmonale neoplasmer. De fleste pulmonale hamartomer diagnosticeres tilfældigt hos patienter i alderen 40 til 70 år, men er meget sjældne i pædiatrisk praksis.
Generelt diagnosticeres de fleste hamartomer hos mænd, selvom de i nyrerne er mere almindelige hos kvinder og identificeres i middelalderen.
Omkring 5% af godartede brysttumorer er hamartomer, og de rammer oftest kvinder over 35 år.
80-90% af hamartomatøse læsioner i hjernen og mere end 50% af hamartomer i hjertet er forbundet med tuberøs sklerose.
Årsager hamartomer
Gamartomer tilhører medfødte misdannelser og er formationer af godartet karakter, som dannes af mesenkymale væv, der stammer fra germinallag. Og årsagerne til deres forekomst er forbundet med ukontrolleret celledeling af cytologisk normalt væv (bindevæv, glat muskulatur, fedt eller brusk), karakteristisk for en given anatomisk placering, og deres fokale overvækst under embryogenesen af næsten ethvert organ eller anatomisk struktur.
Forekomsten af flere hamartomer hos den samme patient omtales ofte som hamartomatose eller pleiotropisk hamartom.
Disse tumorer kan forekomme sporadisk eller i nærvær af visse autosomalt dominante arvelige sygdomme samt genetisk bestemte syndromer.
I mange tilfælde dannes hamartomer, når en sjælden genetisk sygdom af multisystemisk natur - tuberøs sklerose - manifesterer sig kort efter fødslen, eller ved Recklinghausens familiære sygdom - neurofibromatose type 1. [ 2 ]
Risikofaktorer
De vigtigste risikofaktorer for dannelse af hamartomer inkluderer tilstedeværelsen af såkaldte genetiske syndromer af hamartomatøs polypose i patientens historie, herunder:
- Multipelt hamartomsyndrom - Cowden syndrom, hvor der dannes flere hamartomer af ekto-, ento- og mesodermal oprindelse, observeres gastrointestinal polypose og mukokutane manifestationer;
- Peutz-Jeghers-Turen syndrom (karakteriseret ved udvikling af godartede hamartomatøse polypper i mave-tarmkanalen);
- Proteus syndrom;
- Weil syndrom - juvenil polypose i tyktarmen;
- Bannayan-Riley-Ruvalcaba syndrom, som ligesom Cowden syndrom producerer flere hamartomer (hamartomatøse polypper) i tarmen;
- Carney-Stratakis syndrom og Carney-kompleks.
Derudover dannes hamartomer hos patienter med arveligt Watson syndrom og i tilfælde af sporadisk forekommende eller medfødt Pallister-Hall syndrom med hypothalamisk hamartom og polydaktyli.
Patogenese
Mekanismen for øget proliferation af germinalvæv med dannelsen af tumorlignende misdannelser i forskellige organer forklares af kromosomafvigelser og genmutationer, der kan forekomme spontant eller være arvelige.
Ved tuberøs sklerose er der identificeret mutationer i TSC1- eller TSC2-generne - tumorsuppressorer, der forhindrer og hæmmer overdreven proliferation - for hurtig eller ukontrolleret cellevækst og -deling. Og ved neurofibromatose type 1 og Watson syndrom - kimlinjemutationer i det mitokondrielle tumorsuppressorgen NF1.
Ved hamartom-tumorsyndrom, som kombinerer Cowden-, Protea-, Bannayan-Riley-Ruvalcaba- og juvenil polyposissyndrom, er patogenesen forbundet med en mutation af PTEN-genet, som koder for et enzym involveret i reguleringen af proliferation og betragtes som et tumorsuppressorgen.
Mutationer i STK11-genet, der koder for strukturen og funktionen af et af de transmembrane serinenzymer, hvilket reducerer dets evne til at begrænse celledeling, fører til Peutz-Jeghers-Turen syndrom med udvikling af tarmpolypper og pigmenterede hudlæsioner. En mutation i GLI3-genet, en transkriptionsfaktor involveret i dannelsen af intrauterin væv, er blevet identificeret ved Pallister-Hall syndrom.
Således fører ukontrolleret cellevækst på grund af genmutationer til dannelse af hamartom.
Symptomer hamartomer
Afhængigt af lokaliseringen af hamartomer skelnes deres typer, og hver af dem har sin egen struktur og symptomatologi.
Hamartom i lungen
Pulmonalt hamartom kan dannes i enhver lap og perifere dele af lungerne og består af normalt væv i lungerne: fedtvæv, epitelvæv, fibrøst væv og bruskvæv. I 80% af tilfældene dominerer den chondroide komponent (hyaline bruskceller) med inklusion af adipocytter - fedtvævsceller og luftvejsepitelceller. [ 3 ]
De tidligere navne: chondroid hamartom, mesenchymom, chondromatøst hamartom eller hamartochondroma anbefales i øjeblikket ikke af WHO.
Mesenkymalt cystisk hamartom i lungen er derimod mindre almindeligt og er forbundet med Cowden syndrom hos de fleste patienter.
Hamartomatøs læsion i lungen manifesterer sig muligvis ikke selv, men kan forårsage symptomer i form af kronisk hoste (ofte med hæmoptyse), hvæsen ved vejrtrækning og vejrtrækningsbesvær. [ 4 ]
Et hamartom i hjertet
Godartede primære hjertetumorer hos voksne omfatter modent myocythamartom, og hos spædbørn og børn med tuberøs sklerose, rhabdomyom, dvs. myokardiehamartom i ventriklerne eller interventrikulær septum. [ 5 ]
Modent kardiomyocyt-hamartom udvikler sig i ventriklavggen (og sjældent i atrierne) og kan fremstå som flere læsioner, der er tætte masser tæt forbundet med det underliggende myokardium. Tumoren kan forårsage symptomer på hjertesvigt: brystsmerter, hjertebanken og arytmier, hjertemislyde, ødem, dyspnø, cyanose.
Hjerterabdomyomer, hvoraf de fleste diagnosticeres inden for det første leveår, består af hjertemuskelvæv dannet af embryonale myoblaster og har udseende af faste fokale masser uden kapsel.
Typisk præsenterer disse hamartomer sig asymptomatisk og forsvinder spontant før 4-årsalderen.
Hamartomatøse læsioner anses også af nogle eksperter for at være forbundet med Carney-kompleksets myxom i hjertet. [ 6 ]
Gamartom i mave-tarmkanalen
Gastrisk hamartom er en mesenkymal masse i form af en epitelial hyperplastisk polyp i maven, Peutz-Jeghers polyp og et sjældent myoepitelialt hamartom - med hypertrofierede glatmuskelbundter. Andre navne for dette hamartom inkluderer myoglandulært hamartom, adenomyomatøst hamartom og gastrisk adenomyom. Typiske kliniske manifestationer inkluderer dyspepsi, epigastriske smerter og blødning fra øvre mave-tarmkanal. [ 7 ], [ 8 ]
Mere information i materialet - gastrisk polypose
Et intestinalt hamartom er en hamartomatøs eller hyperplastisk polyp i tyktarmen, diagnosticeret som et adenomatøst eller tubulært adenom. Når hamartomet er lokaliseret i Brunner-kirtlen i tolvfingertarmen, manifesterer symptomerne sig ved smerter i den epigastriske region; kvalme, opkastning og flatulens (hvilket indikerer tarmobstruktion); og, hvis det er af betydelig størrelse, gastrointestinal blødning. I tilfælde af myoepitelialt hamartom i ileum klager patienterne over mavesmerter, har nedsat kropsvægt og udvikler kronisk anæmi. [ 9 ], [ 10 ]
Læs også - rektale polypper
Retrorektalt hamartom er et cystisk hamartom eller en flerkammercyste i det retrorektale rum (det løse bindevæv mellem endetarmen og dens egen fascia), der oftest forekommer hos midaldrende kvinder. Det ligner en cyste, der buler ud af endetarmens bagvæg, som er foret med epitel og indeholder kaotisk arrangerede glatte muskelfibre. Dette hamartom viser sig med smerter i den nedre del af maven og tilbagevendende forstoppelse. [ 11 ], [ 12 ]
Hamartomer i leveren og milten
Multipelt biliært hamartom i leveren er et hamartom i de interdollice intrahepatiske galdegange forbundet med misdannelser i deres udvikling i den embryonale periode. Dette hamartom (enkelt eller multipelt) består af tilfældigt udvidede klynger af galdegange og fibrokollagenøst stroma. [ 13 ]
Biliære hamartomer er asymptomatiske og opdages normalt tilfældigt (under radiologisk undersøgelse eller laparotomi). [ 14 ]
En sjælden og ofte tilfældigt opdaget primær neoplasme af godartet karakter er et hamartom i milten, som består af elementer af miltens røde pulpa - i form af en veldefineret homogen masse med fast konsistens. Denne misdannelse kan være enkelt eller flerdobbelt; når man klemmer miltparenkym, kan der være en følelse af ubehag og smerte i venstre subkostalområde. [ 15 ], [ 16 ]
Renale hamartomer
Det mest almindelige hamartom i nyren diagnosticeres som angiomyolipom i nyren, da denne godartede tumor består af modent fedtvæv med indlejrede glatte muskelfibre og blodkar. Den dannes ved tuberøs sklerose i 40-80% af tilfældene. Forøgelse af hamartomets størrelse (mere end 4-5 cm) fører til smerter og forekomst af blod i urinen. [ 17 ], [ 18 ]
Hamartom i brystet
De WHO-accepterede diagnostiske definitioner af brysthamartom er termer som adenolipoma, kondrolipoma og myoidhamartom. Selvom mammologer ofte kalder det fibroadenolipoma, fordi tumorformationen indeholder celler af fibrøst, kirtel- og fedtvæv indesluttet i en tynd bindevævskapsel med tydelige konturer. Fokale forkalkninger kan observeres ved visualisering. I dette tilfælde er kliniske manifestationer fraværende. [ 19 ], [ 20 ]
Læs også - brysttumorer
Hamartomer i hjernen
En tredjedel af patienter med tuberøs sklerose har et hjernehamartom i form af intrakranielle kortikale udvækster eller tuberkler i forskellige lapper - ved grænsen mellem grå og hvid substans - eller subependymale knuder langs væggene i hjerneventriklerne. Astrocytisk hamartom, et subependymalt kæmpecelle-astrocytom med kortikal forstyrrelse, dysmorfe neuroner og store gliaceller i hjerneparenkym (astrocytter), kan også dannes. Symptomer på cerebrale hamartomer omfatter anfald og mental retardering hos børn. [ 21 ], [ 22 ]
En sjælden misdannelse, der opstår under embryogenesen og er til stede ved fødslen, er et hypothalamisk hamartom, som er en masse af heterotopiske neuroner og gliaceller. Efterhånden som barnets hjerne vokser, forstørres tumoren, men spreder sig ikke til andre hjerneområder. [ 23 ], [ 24 ]
Hvis der dannes hypertrofieret væv i den forreste del af hypothalamus (tuber cinereum), hvor hypofysen hæfter sig til den, viser misdannelsen symptomer på central for tidlig seksuel udvikling (før 8-9 år): forekomst af akneudslæt, tidlig udvikling af brystkirtler og tidlig menarche hos piger; tidlig kønsbehåring og stemmemutation hos drenge.
Når hamartomer dannes i den bageste del af hypothalamus, kan der være abnormiteter i hjernens elektriske aktivitet, hvilket i den tidlige barndom manifesterer sig ved anfald og på et senere tidspunkt (4 til 7 år) ved epilepsi med fokale epileptiske anfald med pludselig latter eller ufrivillig gråd, atoniske og tonisk-kloniske anfald samt aggressionsanfald, hukommelses- og kognitive problemer.
Et hypofysehamartom er et sporadisk forekommende godartet hypofyseadenom.
Middelaldrende voksne med Cowden syndrom kan have en sjælden tumorlignende masse, et hamartom i lillehjernen, diagnosticeret som dysplastisk cerebellært gangliocytom eller Lhermitte-Duclos sygdom. Symptomer kan være fraværende eller manifestere sig som hovedpine, svimmelhed, nedsat koordination af bevægelser og lammelse af individuelle kranienerver.
Lymfeknudehamartom
Når cellerne i glat muskulatur og fedtvæv, såvel som blodkar og kollagent stroma i inguinale, retroperitoneale, submandibulære og cervikale lymfeknuder, vokser over, dannes et angiomyomatøst hamartom i en lymfeknude eller et nodulært angiomyomatøst hamartom - med delvis eller fuldstændig udskiftning af dens parenkym. [ 25 ], [ 26 ]
Et hamartom i huden
I nærvær af tuberøs sklerose eller neurofibromatose observeres forskellige hudhamartomer, oftest i form af hypopigmenterede pletter; kaffe- og mælkepletter; angiofibrom (på kinderne, hagen, nasolabiale folder); shagreen-pletter af forskellige lokaliseringer (som er bindevævsnævi); fibrøse plaques på panden, hovedbunden eller halsen.
En sjælden dermatologisk manifestation af tuberøs sklerose (især hos mænd) er follikulocystisk og kollagen hamartom, karakteriseret ved rigelig kollagenaflejring i dermis, koncentrisk perifollikulær fibrose og keratinfyldte tragtformede subkutane cyster set ved histopatologisk undersøgelse. [ 27 ]
Med hamartomer bestående af melanocytter (celler, der producerer pigmentet melanin), henviser de fleste eksperter også til forskellige melanocytiske neoplasmer, især medfødte melanocytiske nævi, som repræsenterer en abnormalitet i embryogenesen.
Med hensyn til ætiologi er hamartomer bestående af vaskulært væv også hæmangiomer i huden.
Patienter med Peutz-Jeghers-Thuren syndrom har hamartom i form af pletvis pigmentering af hud og slimhinder - lentiginosis periorificialis
Tilfælde af lineært papuløst ektodermalt-mesodermalt hamartom (Hamartoma moniliformis) viser et lineært, kødfarvet papuløst udslæt på hoved, hals og øvre del af brystet.
Og et sebocytisk hamartom er et hamartom i talgkirtlerne, læs mere i publikationen - talgnævus.
Hamartom i øjet
Pigmenterede hamartomatøse læsioner i iris ved neurofibromatose type 1 og Watson syndrom - i form af nodulære klynger af dendritiske melanocytter - defineres som irishamartomer eller Lisch-knuder. De er transparente (normalt ikke synspåvirkende), afrundede kuppelformede gulbrune papler, der stikker ud over irisoverfladen.
Og patienter med juvenilt angiofibrom i nasopharynx og familiær adenomatøs polypose udvikler ofte et kombineret hamartom i nethinden og retinalt pigmentepitel - i form af en sort plet på den centrale (makulære) del af nethinden. [ 28 ]
Et hamartom i næsen
Nasal hamartom defineres af specialister som nasalt kondromenkymalt hamartom eller nasalt kondrom, der skyldes godartet proliferation af respiratorisk epitel, submukosale kirtler og kondro-knoglemesenkym. Dets kliniske manifestationer afhænger af læsionens størrelse og lokalisering og omfatter: tilstoppet næse, vanskeligheder med nasal vejrtrækning og amning hos spædbørn, klar, vandig næseflåd og næseblødning. Et hamartom kan vokse med barnet og sprede sig ind i øjenhulerne, hvilket resulterer i fremadrettet eller bagudrettet forskydning af øjeæblet, strabismus eller okulomotoriske forstyrrelser. [ 29 ]
Et hamartom hos et barn
Alle de ovennævnte hamartomatøse læsioner i forskellige organer og anatomiske strukturer er til stede hos børn med tilsvarende syndromer.
Nyfødte præsenterer med mesenkymalt hamartom i brystvæggen eller bruskagtigt hamartom i ribbenet, som er faste, immobile masser som følge af fokal overvækst af normale skeletelementer med bruskagtige, vaskulære og mesenkymale elementer. Dette hamartom kan forårsage respirationssvigt og udvikling af respiratorisk distress syndrom. Mesenkymalt hamartom i leveren er den næsthyppigste godartede levertumor hos børn. Denne tumorlignende formation (oftere lokaliseret i organets højre lap) består af celler fra mesenkymalt stroma, hepatocytter og epitelceller i galdevejsforingen. Det kliniske billede omfatter en palpabel masse i bughulen, anoreksi og vægttab, og i tilfælde af betydelig størrelse (op til 10 cm og mere) dækker tumoren ekstrahepatiske galdeveje og vena cava inferior, hvilket fører til gulsot og ødem i underekstremiteterne.
Et hamartom er et medfødt mesoblastisk nefrom (forekommer hos 1 ud af 200.000 spædbørn), der kan resultere i oppustethed i maven hos den nyfødte med en palpabel masse af tæt konsistens i den øvre højre kvadrant af maven. Spædbørn kan også have hurtig, overfladisk vejrtrækning.
Sjældne medfødte anomalier omfatter fibrøst hamartom i spædbarnsalderen, som forekommer hos børn i de første to leveår og præsenterer sig som en smertefri nodulær masse i det subkutane væv i armhulen, nakken, skulderen og underarmen, ryggen og brystet, låret, fødderne og de ydre kønsorganer.
Ekkrin angiomatøs hamartom hos et barn kan være til stede ved fødslen eller manifestere sig i den tidlige barndom. Denne godartede tumor af hamartomatøs natur har normalt udseendet af blålige eller brunlige knuder og/eller plaques, der skyldes proliferation af ekkrin svedkirtelvæv og kapillærer i de midterste og dybe lag af dermis. Dette hamartom kan forårsage lokaliseret hyperhidrose og øget hårvækst.
Komplikationer og konsekvenser
Det er almindelig enighed om, at hamartomer sjældent vender tilbage eller omdannes til ondartede tumorer. De viser ofte få eller ingen symptomer og forsvinder nogle gange endda med tiden. Men i mere alvorlige tilfælde og afhængigt af dannelsesstedet kan disse misdannelser have alvorlige komplikationer og konsekvenser.
Først og fremmest kan et hamartom vokse til en sådan størrelse, at det begynder at presse på omgivende væv og organer og forstyrrer deres funktioner.
Hjertehamartom hos børn kan føre til vedvarende hjerterytmeforstyrrelser, klapdefekter og nedsat intrakardial blodgennemstrømning med efterfølgende kongestiv hjertesvigt.
Komplikationer ved hamartomatøse polypper i mave-tarmkanalen er gastrointestinal blødning, obstruktion og intestinal invagination (med mulig dødelig udgang). Og et stort renalt hamartom kan fremkalde nyreruptur.
Et hamartom i hjernen kan forårsage obstruktiv hydrocephalus syndrom.
Ved hypothalamiske og hypofysehamartomer kan produktionen af somatotropisk hormon (væksthormon) være nedsat, hvilket kan føre til udvikling af hypofyseal nanisme (hypopituitarisme) hos børn. Hypothalamiske hamartomer hos børn kan også føre til lægemiddelresistent epilepsi.
Komplikationer ved retinalt pigmentepitelhamartom er fyldt med nethinde- og/eller synsnervedysfunktion, makulaødem, neovaskularisering af årehinden og nethindeløsning.
Diagnosticering hamartomer
En vigtig del af diagnostikken af hamartomer og relaterede syndromer er indsamling af anamnese, herunder familiehistorie.
Laboratorieundersøgelser omfatter blodprøver: generelle kliniske prøver; serumelektrolytter; lymfocytprofil; calcium-, kalium-, fosfat- og urinstofniveauer; og leverfunktionstest. Hvis det er muligt, udføres en finnålsaspirationsbiopsi af massen, da histologisk undersøgelse er afgørende for diagnosen og valget af behandlingstaktik.
Instrumentel diagnostik giver visualisering af hamartomatøs tumorlignende dannelse og identifikation af dens nøjagtige lokalisering, hvortil røntgen, angiografi, elektroencefalografi (EEG), ultralyd (sonografi), CT (computertomografi), PET (positronemissionstomografi) og MR (magnetisk resonansbilleddannelse) anvendes.
Differential diagnose
Ved alle unormale masser er differentialdiagnose meget vigtig. Derfor differentieres der mellem tuberkulom og hamartom; pulmonært hamartom og primær lungekræft, bronkogen karcinoid sygdom, metastatisk sygdom. Hjernehamartom bør skelnes fra kraniofaryngeom og hypothalamisk-kiasmatisk gliom. Og differentialdiagnosen af hamartom som medfødt mesoblastisk nefrom inkluderer Wilms tumor (malignt nefroblastom), klarcellet sarkom i nyrerne og ossificerende nyretumor hos spædbørn.
Hvem skal kontakte?
Behandling hamartomer
Hvis hamartomet er asymptomatisk og opdages ved et uheld, er behandling ikke nødvendig, men det er nødvendigt at overvåge dets "adfærd" og patientens tilstand. I andre tilfælde sigter terapien mod at reducere symptomernes intensitet og forebygge komplikationer. For eksempel ordineres der ved hypothalamisk hamartom med symptomer på for tidlig pubertet visse lægemidler, der hæmmer frigivelsen af bestemte hormoner. Hjertemedicin bruges til at behandle symptomer på hjertesvigt hos patienter med hjertehamartomer.
Kirurgisk fjernelse af hamartomer er indiceret for at bekræfte diagnosen og i tilfælde af medicinsk ukorrigerbare intense symptomer.
For eksempel kan lungehamartomer fjernes ved kileresektion og i alvorlige tilfælde ved fjernelse af en lungelap (lobektomi). Et brysthamartom kan også fjernes, og hvis det er stort, kan en delvis eller fuldstændig mastektomi være nødvendig.
Stereotaktisk radiofrekvenstermoablation eller laserablation kan anvendes til at fjerne hamartomatøse polypper. Radiokirurgi med stærkt fokuserede gammastråler - gammakniv til hypothalamiske hamartomer eller astrocytiske hamartomer - anvendes også.
Forebyggelse
Den eneste metode til at forhindre udviklingen af hamartomer kan betragtes som genetisk screening af barnets fremtidige forældre.
Vejrudsigt
Den samlede prognose for denne medfødte anomali afhænger af lokaliseringen og størrelsen af neoplasmen, samt af komorbiditeter og patientens generelle helbredstilstand.