Medicinsk ekspert af artiklen

Nye publikationer
Prions - forårsagende agens af prionsygdomme
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Langsom virusinfektioner er kendetegnet ved særlige kriterier:
- usædvanlig lang inkubationsperiode (måneder, år)
- en slags nederlag af organer og væv, hovedsageligt centralnervesystemet;
- langsom, stabil fremgang af sygdommen
- et uundgåeligt dødeligt udfald.

Nogle patogener af akutte virale infektioner kan også forårsage langsomme virale infektioner. For eksempel forårsager mæslingsvirus i nogle tilfælde PSES og rubella-viruset - progressiv medfødt røde hunde og rubella-panencephalitis.
En typisk langsom viral infektion af dyr er forårsaget af virus visna / medi, som tilhører retrovirus. Det er forårsagende middel til langsom viral infektion og progressiv lungebetændelse hos får. Den hvide substans i hjernen er ødelagt , forlamning udvikler sig (visna - miste); der er en kronisk betændelse i lungerne og milten.
Lignende sygdomme på grund af langsom virusinfektioner forårsager prioner - patogener af prioninfektioner. Prionsygdomme er en gruppe af progressive forstyrrelser i humane og dyre CNS. Folk har forstyrret funktionen af centralnervesystemet, der er ændringer i personlighed, bevægelsesforstyrrelser. Symptomer på sygdommen varer normalt fra flere måneder til flere år og slutter dødelig. Tidligere blev prioninfektioner betragtet sammen med de såkaldte patogener af langsom virusinfektioner.
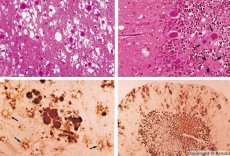
Nogle agenter, der forårsager prionsygdomme, akkumuleres først i lymfoide væv. Prioner ind i hjernen, det ophobes i store mængder, der forårsager amyloidose (ekstracellulær disproteinoz kendetegnet ved amyloiddeponering med udvikling af atrofi og sklerose af væv) og astrocytose (astrocytisk glia proliferation, giperprodukdiya glialnyh fibre). Fibriller eller amyloid proteinaggregater og hjerne spongiform ændring (overførbar spongiform encephalopati). Som et resultat, ændringer adfærd forstyrret koordinering af bevægelser, der er udviklet fatal udtynding. Immunitet er ikke dannet. Prion sygdomme relateret til konformationelle sygdomme, der udvikles som et resultat af misfoldning (krænkelse af korrekte konformation) af cellulært protein, der kræves for normal krop fungerer. Manierne til overførsel af prioner er forskellige:
- spiserøret - inficerede produkter af animalsk oprindelse, kosttilskud fra råkvægorganer mv:
- overførsel med blodtransfusion, administration af animalske produkter, transplantation af organer og væv, anvendelse af inficerede kirurgiske og dentalinstrumenter;
- transmission via immunobiologiske lægemidler (kendt for at inficere PrpP '' '1500 får hjerne formulvaccine fra syge får).
Patologiske prioner, der er kommet ind i tarmene, transporteres til blod og lymfe. Efter perifer replikation i milten, appendiks, mandler og andre lymfoide væv overføres de til hjernen via perifere nerver (neuroinvasi). Sandsynligvis direkte indtrængning af prioner i hjernen gennem blod-hjernebarrieren. Tidligere blev det antaget, at centralnervesystemet er det eneste væv, hvor patologiske prioner akkumuleres, men undersøgelser er opstået, der ændrede denne hypotese. Det viste sig, at akkumuleringen af prioner i milten er forbundet med stigningen og funktionen af follikulære dendritiske celler.
 [
[Egenskaber af prioner
Den cellulære normale isoform af prionprotein med en molekylvægt på 33-35 kD bestemmes af prionproteegenet (priongen-PrNP er på det 20. Kromosom hos et menneske). Et normalt gen fremkommer på overfladen af cellen (forankret i membranen med glycoproteinmolekylet), er følsomt for proteasen. Det regulerer transmissionen af nerveimpulser, døgncykler, oxidationsprocesser, deltager i metabolismen af kobber i centralnervesystemet og i reguleringen af opdeling af knoglemarvstamceller. Desuden findes prion genet i milt, lymfeknuder, hud, GIT og follikulære dendritiske celler.
Spredning af patologiske prioner
Omdannelsen af prioner til ændrede former opstår, når den kinetisk kontrollerede ligevægt mellem dem er forstyrret. Processen forbedres, når mængden af patologisk (PrF) eller eksogent prion stiger. PrP er et normalt protein forankret i cellemembranen. PrP 'er et kugleformet hydrofobt protein, som danner aggregater med sig selv og med PrF "på overfladen af cellen: Som et resultat omdannes PrP til PrF" og cyklussen fortsætter. Den patologiske form af PrF "" akkumuleres i neuroner, hvilket giver cellen en svampet udseende.
Kuru
Prionsygdom, der tidligere var udbredt blandt papuanserne (oversat som rysten eller rysten) i den østlige del af øen New Guinea. Infektiøse egenskaber af sygdommen viste K. Gaidushek. Den forårsagende middel overføres af fødevarer som følge af rituelle kannibalisme - spiser utilstrækkeligt termisk behandlede inficerede prioner af hjernedødsledte. Som et resultat af nederlaget i centralnervesystemet forstyrres bevægelser, gang, rystelse, eufori ("latterlig død"). Inkubationsperioden varer 5-30 år. Et år senere dør patienten.
Creutzfeldt-Jakob sygdom
Prionsygdom, som strømmer i form af demens, visuel og cerebellare lidelser og bevægelseslidelser dødelig sygdom i 4-5 måneder i den klassiske variant Creutzfeldt-Jakobs sygdom og i (3-14 måneder, når nye variant af Creutzfeldt-Jakobs sygdom. Inkubationstiden kan nå 20. Der er forskellige måder at inficere på og årsagerne til sygdommen:
- når der anvendes utilstrækkeligt varmebehandlede produkter af animalsk oprindelse, fx kød, hjernekøer, patienter med bovin spongiform encephalopati;
- vævstransplantation, fx corneal, blodtransfusion, anvendelse af hormoner og andre biologisk aktive stoffer af animalsk oprindelse, anvendelse af catgut eller utilstrækkeligt prosterilinovannyh kontaminerede kirurgiske instrumenter, manipulation dissekere;
- til hyperproduktion af PrP og andre stater, der stimulerer processen med transformation af PrP 'til PrF. "
Sygdommen kan også udvikle som et resultat af en mutation eller insertion i prion-genregionen. Familiens karakter af sygdommen er udbredt som et resultat af en genetisk prædisponering for Creutzfeldt-Jakobs sygdom. Med en ny variant af Creutzfeldt-Jakobs sygdom udvikles sygdomme i en yngre alder (middelalder 28 år) i modsætning til den klassiske variant (middelalder 65 år). Med den nye variant af Creutzfeldt-Jakobs sygdom akkumuleres unormalt prionisk protein ikke kun i centralnervesystemet, men også i lymfektetiske væv, herunder i tonsiller.
Gerstmann-Streussler-Sheinker-syndromet
Arvelig prionsygdom, der opstår med demens, hypotension, synke (dysfagi), dysartri. Ofte bærer en familie karakter. Inkubationsperioden er fra 5 til 30 år. Sygdommen opstår i 50-60 år, dens varighed varierer fra 5 til 13 år.
Arvelig dødelig søvnløshed
Autoimmun sygdom med progressiv søvnløshed, sympatisk hyperresponsivitet (hypertension, pyreksi, udslæt, takykardi), tremor, ataksi, mnokloniyami, hallucinationer. Søvn er stærkt forstyrret. Døden opstår med progression af kardiovaskulær svigt.
Løs det
Scrapie (fra det engelske skrabe -. Skrab) - prionsygdom af får og geder (many), flyder med CNS sygdom, progressiv sygdom i bevægelse, stærk pruritus (kløe), og ender i dyrets død.
Bovin spongiform encephalopati
Sygdom af kvæg, der er karakteriseret ved nederlaget i centralnervesystemet, en krænkelse af koordinationen af bevægelser og dyrets uundgåelige død. For første gang brød epidemien ud af sygdommen i Storbritannien. Det var forbundet med fodring af dyr med kød- og benmel indeholdende patologiske prioner. Inkubationsperioden varierer fra 1,5 til 15 år. De mest inficerede er hoved, rygmarv og øjne af dyr.
Laboratoriediagnostik af prionsygdomme
Ved diagnosticering, spongiforme ændringer i hjernen, astrocytose (gliose), fravær af infiltrater af inflammation. Hjernen er farvet med amyloid. I cerebrospinalvæsken påvises proteinmarkører af prionhjerneforstyrrelser (ved hjælp af ELISA). Genetisk analyse af priongen (PCR) udføres.
Profylakse af prionsygdomme
Til dekontaminering af værktøjer og miljø objekter anbefalet af autoklavering (ved 134 ° C 18 min ved 121 ° C i 1 time)., Burning, yderligere blegemiddel behandling og odnonormalnym opløsning af NaCl i 1 time for ikke-specifik forebyggelse lægger begrænsninger på anvendelsen af lægemidler af animalsk oprindelse og Produktionen af hypofysehormoner af animalsk oprindelse er forbudt. Begræns transplantationen af dura mater. Ved arbejde med de dialogiske væsker af patienter, der bruger gummihandsker.