Medicinsk ekspert af artiklen

Nye publikationer
Prioner - årsag til prionsygdomme
Sidst revideret: 06.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Langsomme virusinfektioner er karakteriseret ved særlige kriterier:
- en usædvanlig lang inkubationsperiode (måneder, år);
- en specifik læsion af organer og væv, primært centralnervesystemet;
- langsom, stabil progression af sygdommen;
- uundgåelig fatal udgang.

Nogle patogener, der forårsager akutte virusinfektioner, kan også forårsage langsomme virusinfektioner. For eksempel forårsager mæslingevirus undertiden SSPE, og røde hundevirus forårsager progressiv medfødt røde hunde og røde hunde panencephalitis.
En typisk langsom virusinfektion hos dyr er forårsaget af visna/madi-virus, som er en retrovirus. Det er årsagen til langsom virusinfektion og progressiv lungebetændelse hos får. Hjernens hvide substans ødelægges, der udvikles lammelse (visna - henfald); kronisk betændelse i lunger og milt opstår.
Sygdomme, der i deres træk ligner langsomme virusinfektioner, er forårsaget af prioner - de forårsagende agenser for prioninfektioner. Prionsygdomme er en gruppe af progressive lidelser i centralnervesystemet hos mennesker og dyr. Hos mennesker er centralnervesystemets funktion nedsat, der forekommer personlighedsændringer, og der opstår bevægelsesforstyrrelser. Symptomerne på sygdommen varer normalt fra flere måneder til flere år og ender med døden. Tidligere blev prioninfektioner betragtet sammen med de såkaldte forårsagende agenser for langsomme virusinfektioner.
Nogle stoffer, der forårsager prionsygdomme, akkumuleres først i lymfoidvæv. Prioner, der trænger ind i hjernen, akkumuleres i store mængder, hvilket forårsager amyloidose (ekstracellulær dysproteinose, karakteriseret ved aflejring af amyloid med udvikling af atrofi og sklerose i vævet) og astrocytose (proliferation af astrocytiske neuroglia, hyperproduktion af glialfibre). Fibriller, aggregater af protein eller amyloid og svampeforme forandringer i hjernen (overførbare svampeforme encephalopatier) dannes. Som følge heraf ændres adfærd, koordinationen af bevægelser forringes, og der udvikles udmattelse med dødelig udgang. Immunitet dannes ikke. Prionsygdomme er konformationssygdomme, der udvikles som følge af forkert foldning (krænkelse af den korrekte konformation) af cellulært protein, der er nødvendigt for kroppens normale funktion. Ruterne for prions transmission er varierede:
- fordøjelsesvej - inficerede produkter af animalsk oprindelse, fødevaretilsætningsstoffer fra rå kvægorganer osv.:
- smitte via blodtransfusion, administration af lægemidler af animalsk oprindelse, organ- og vævstransplantation, brug af inficerede kirurgiske og dentale instrumenter;
- transmission gennem immunbiologiske præparater (infektion af 1500 får med PrP''' med hjerneformolvaccine fra syge får er kendt).
Patologiske prioner, der er kommet ind i tarmen, transporteres ind i blodet og lymfen. Efter perifer replikation i milten, blindtarmen, mandlerne og andet lymfoidt væv overføres de til hjernen gennem de perifere nerver (neuroinvasion). Direkte penetration af prioner ind i hjernen gennem blod-hjerne-barrieren er mulig. Tidligere troede man, at centralnervesystemet var det eneste væv, hvor patologiske prioner akkumuleres, men der er dukket undersøgelser op, der har ændret denne hypotese. Det viste sig, at akkumulering af prioner i milten er forbundet med stigningen og funktionen af follikulære dendritiske celler.
 [
[ Prioners egenskaber
Den normale cellulære isoform af prionproteinet med en molekylvægt på 33-35 kDa bestemmes af prionproteingenet (priongenet - PrNP er placeret på det 20. menneskelige kromosom). Det normale gen optræder på celleoverfladen (forankret i membranen af molekylets glykoprotein), følsomt over for protease. Det regulerer transmissionen af nerveimpulser, daglige cyklusser, oxidationsprocesser, deltager i kobbermetabolisme i centralnervesystemet og i reguleringen af knoglemarvsstamcelledeling. Derudover findes priongenet i milten, lymfeknuder, hud, mave-tarmkanal og follikulære dendritiske celler.
Spredning af patologiske prioner
Transformationen af prioner til ændrede former sker, når den kinetisk kontrollerede ligevægt mellem dem forstyrres. Processen forstærkes af en stigning i mængden af patologisk (PrP) eller eksogent prion. PrP er et normalt protein forankret i cellemembranen. PrP' er et globulært hydrofobt protein, der danner aggregater med sig selv og PrP'' på celleoverfladen: som et resultat transformeres PrP' til PrP'', og derefter fortsætter cyklussen. Den patologiske form af PrP''' akkumuleres i neuroner, hvilket giver cellen et svampet udseende.
Kuru
Prionsygdom, tidligere almindelig blandt papuanere (som betyder rysten eller rysten) i den østlige del af øen Ny Guinea. Sygdommens smitsomme egenskaber blev bevist af K. Gajdusek. Patogenet overføres via mad som følge af rituel kannibalisme - at spise den utilstrækkeligt tilberedte, prioninficerede hjerne fra afdøde slægtninge. Som følge af skader på centralnervesystemet forringes bevægelse og gang, kulderystelser og eufori ("latterdød") opstår. Inkubationsperioden varer 5-30 år. Patienten dør efter et år.
Creutzfeldt-Jakobs sygdom
Prionsygdom, som manifesterer sig som demens, syns- og cerebellare forstyrrelser og bevægelsesforstyrrelser med dødelig udgang efter 4-5 måneders sygdom ved den klassiske variant af Creutzfeldt-Jakobs sygdom og efter (3-14 måneder ved den nye variant af Creutzfeldt-Jakobs sygdom. Inkubationstiden kan nå op på 20 år. Forskellige smitteveje og årsager til sygdommen er mulige:
- ved indtagelse af utilstrækkeligt varmebehandlede animalske produkter, såsom kød og hjerner fra køer med bovin spongiform encephalopati;
- under vævstransplantation, såsom hornhindetransplantation, blodtransfusion, brug af hormoner og andre biologisk aktive stoffer af animalsk oprindelse, brug af catgut, kontaminerede eller utilstrækkeligt steriliserede kirurgiske instrumenter, prosektorale manipulationer;
- i tilfælde af hyperproduktion af PrR og andre tilstande, der stimulerer processen med at omdanne PrR' til PrR".
Sygdommen kan også udvikle sig som følge af en mutation eller indsættelse i prion-genregionen. Sygdommens familiære natur er almindelig på grund af genetisk prædisposition for Creutzfeldt-Jakobs sygdom. I den nye variant af Creutzfeldt-Jakobs sygdom udvikles lidelserne i en yngre alder (gennemsnitsalder 28 år) i modsætning til den klassiske variant (gennemsnitsalder 65 år). I den nye variant af Creutzfeldt-Jakobs sygdom akkumuleres unormalt prionprotein ikke kun i centralnervesystemet, men også i lymforetikulært væv, herunder mandlerne.
Gerstmann-Sträussler-Scheinkers syndrom
Arvelig prionsygdom, ledsaget af demens, hypotoni, synkebesvær (dysfagi), dysartri. Har ofte en familiær karakter. Inkubationsperioden er fra 5 til 30 år. Sygdommen opstår i 50-60-årsalderen, og dens varighed varierer fra 5 til 13 år.
Arvelig dødelig søvnløshed
En autoimmun sygdom med progressiv søvnløshed, sympatisk hyperreaktivitet (hypertension, hypertermi, hyperhidrose, takykardi), tremor, ataksi, multiklon, hallucinationer. Søvnen er alvorligt forstyrret. Døden indtræffer ved progression af kardiovaskulær svigt.
Skrabe
Scrapie (fra engelsk scrape - at skrabe) er en prionsygdom hos får og geder (fnat), som opstår med skader på centralnervesystemet, progressive bevægelsesforstyrrelser, svær hudkløe (fnat) og ender med dyrets død.
Bovin spongiform encefalopati
En sygdom hos kvæg, der er karakteriseret ved skader på centralnervesystemet, nedsat koordination af bevægelser og dyrets uundgåelige død. Sygdommens epidemi brød først ud i Storbritannien. Den var forbundet med fodring af dyr med kød- og benmel indeholdende patologiske prioner. Inkubationsperioden varierer fra 1,5 til 15 år. Dyrenes hjerne, rygmarv og øjne er mest inficerede.
Laboratoriediagnostik af prionsygdomme
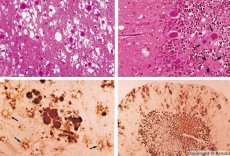
Under diagnostikken observeres svampformede forandringer i hjernen, astrocytose (gliose) og fravær af inflammatoriske infiltrater. Hjernen farves for amyloid. Proteinmarkører for prionsygdomme i hjernen påvises i cerebrospinalvæsken (ved hjælp af ELISA). Genetisk analyse af priongenet (PCR) udføres.
Forebyggelse af prionsygdomme
Autoklavering (ved 134 °C i 18 min; ved 121 °C i 1 time), forbrænding, yderligere behandling med blegemiddel og en 1-N NaCl-opløsning i 1 time anbefales til dekontaminering af instrumenter og miljøgenstande. Til ikke-specifik profylakse er der indført restriktioner for brugen af lægemidler af animalsk oprindelse, og produktion af hypofysehormoner af animalsk oprindelse er forbudt. Transplantation af dura mater er begrænset. Gummihandsker anvendes ved arbejde med patienters dialogvæsker.