Medicinsk ekspert af artiklen

Nye publikationer
Alkaptonuri er en medfødt enzymabnormitet
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
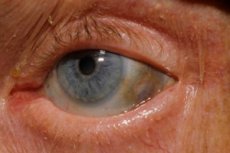
En af de meget sjældne stofskiftesygdomme, alkaptonuri, refererer til medfødte anomalier i metabolismen af aminosyren tyrosin.
Dette syndrom kan også kaldes homogentisatoxidasemangel, homogentisinuri, arvelig ochronose eller sort urinsygdom.[ 1 ]
Epidemiologi
Ifølge statistikker er der ikke mere end ni tilfælde af alkaptonuri pr. 1 million mennesker. Og i de fleste europæiske lande er der ét tilfælde pr. 100-250 tusind levendefødte.
Blandt de europæiske lande er undtagelsen Slovakiet (især den relativt lille nordvestlige region), hvor prævalensen af alkaptonuri er ét tilfælde pr. 19.000 nyfødte. Dette skyldes højst sandsynligt, at blandt de slovakiske romafamilier, der bor der, er graden af indavl (ægteskab mellem fætre og kusiner) den højeste i Europa: 10-14 %. [ 2 ]
Årsager alkaptonuri
De nøjagtige årsager til alkaptonuri, som er en medfødt lidelse i katabolismen (metabolisk nedbrydning) af den aromatiske (homocykliske) α-aminosyre tyrosin, er blevet fastslået: denne type metabolisk lidelse er en konsekvens af homozygote eller sammensatte heterozygote mutationer i et af de tusindvis af gener på kromosom 3, mere præcist HGD-genet på locus 3q21-q23 på kromosomets lange arm. Dette gen koder for nukleotidsekvenserne af leverenzymet homogentisat-1,2-dioxygenase [ 3 ] (også kaldet homogentisinsyreoxidase eller homogentisatoxidase) - et jernholdigt metalloprotein, der er nødvendigt for et af stadierne i tyrosin-nedbrydningen i kroppen. [ 4 ], [ 5 ]
Alkaptonuri er således en defekt i enzymet homogentisat-1,2-dioxygenase, eller mere præcist, resultatet af dets genetisk bestemte mangel eller fuldstændige fravær. [ 6 ]
Da alkaptonuri er en medfødt enzymmangel, nedarves det autosomalt recessivt. Det vil sige, at for at alkaptonuri kan forekomme hos børn, skal begge forældre have et modificeret gen for enzymet, da hver af dem kun giver én kopi af genet ud af to tilgængelige videre til barnet.
Ifølge de seneste data er der mere end to hundrede varianter af modifikation af HGD-genet, og missense-mutationer, translokation og splejsning observeres oftest.
Risikofaktorer
Den eneste risikofaktor for udvikling af denne medfødte enzymopati er dens tilstedeværelse i familiens historie og arv af to modificerede kopier af HGD-genet, hvis forældrene ikke udviser alkaptonuri (risikoen for at overføre anomalien er 25%), eller hvis en af forældrene har denne lidelse. [ 7 ]
Patogenese
Tyrosin spiller en nøglerolle i syntesen af proteiner, produktionen af kromoproteiner – hudpigmentet melanin, samt skjoldbruskkirtelhormoner og katekolamin-neurotransmittere.
Mekanismen til regulering af mængden af tyrosin i celler er meget kompleks, og kroppen normaliserer dets overskydende indhold ved at nedbryde det. Processen med tyrosinkatabolisme er, ligesom alle aromatiske aminosyrer, flertrinnet og foregår i flere faser. Hvert trin i den metaboliske nedbrydning af tyrosin sker med deltagelse af et specifikt enzym og dannelsen af en mellemliggende forbindelse.
Så først nedbrydes aminosyren til para-hydroxyphenylpyruvat, som omdannes til alkapton – 2,5-dihydroxyphenyleddikesyre eller homogentisinsyre. Derefter skulle alkaptonen omdannes til maleeddikesyre, men dette sker ikke. [ 8 ]
Og patogenesen af alkaptonuri består i ophør af biokemiske reaktioner i tyrosinkatabolismen på stadiet af dannelsen af homogentisinsyre: der er simpelthen ikke behov for noget enzym til at nedbryde det – homogentisatoxidase.
Homogentisinsyre bruges ikke af kroppen og kan ophobes ved udskillelse gennem nyrerne. Derudover oxideres den til benzoquinoacetat (benzoquinoneeddikesyre), som ved at binde sig til vævsmolekyler og kropsvæsker danner biopolymerforbindelser farvet som melanin.
Ophobningen af disse mellemprodukter i vævet fører til en forstyrrelse af bruskvævets kollagenstruktur, hvilket reducerer dets elasticitet – med forekomsten af mange kliniske tegn på alkaptonuri og udvikling af komplikationer.
Symptomer alkaptonuri
Alkaptonuri hos nyfødte og spædbørn er karakteriseret ved mørkfarvning af urinen. Når urinen på bleer, bleer og undertøj udsættes for luft, bliver den mørkebrun; dette skyldes ophobning og frigivelse af homogentisinsyre, som oxideres til benzoquinoacetat. [ 9 ]
I mangel af andre symptomer opdages alkaptonuri hos små børn ofte ikke rettidigt, da urinen kan blive mørkere efter flere timers vandladning. Ifølge nogle data identificeres kun en femtedel af børn under 12 måneder, der er født med denne enzymmangel, klinisk. Derfor er det meget vigtigt, at forældre er opmærksomme på at passe på deres spædbørn.
Derudover omfatter tidlige tegn pigmentering (blågrå farve) af øjnenes senehinde og brusken i ørerne og næsen, hvilket ofte kaldes ochronose.[ 10 ]
Over tid opstår andre symptomer:
- alvorlig pigmentering af huden på kindben, armhuler og kønsorganer;
- misfarvning af tøj ved kontakt med svedige områder af kroppen;
- anfald af generel svaghed;
- hæs stemme.
Det skal huskes, at alkaptonuri og ochronose, som nævnt ovenfor, er synonyme navne for den samme lidelse i tyrosinkatabolisme.
Ahornsirup-urinsygdom og alkaptonuri. Medfødt ahornsirup-urinsygdom eller leucinose er også en metabolisk lidelse, har samme arvemønster, og selv mutationer forekommer på samme kromosom, men påvirker genet, der koder for enzymkomplekset af forgrenet α-keto-syredehydrogenase. På grund af dette kan kroppen ikke nedbryde visse komponenter af proteiner, især aminosyrerne leucin, isoleucin og valin. Ved denne sygdom har urin (og ørevoks) en sød lugt; derudover omfatter det kliniske billede af denne type organisk acidæmi hypopigmentering, udsving i blodtrykket, anfald, opkastning og diarré, et fald i blodsukkerniveauet, ketoacidose, hallucinationer osv. Dødeligheden hos børn er ret høj; hos voksne kan koma og død forekomme uden behandling på grund af hjerneødem.
Albinisme og alkaptonuri er kun "forenet" af tyrosin. Albinisme, inklusive okulokutan, er forårsaget af genetiske mutationer, der påvirker produktionen af pigmentet melanin. Medfødte forandringer er observeret i TYR-genet på kromosom 11 (11q14.3), som koder for tyrosinase, et kobberholdigt melanosomenzym, der er nødvendigt for dannelsen af hudpigment baseret på tyrosinmetabolismeprodukter. Denne sygdom er meget mere almindelig end alkaptonuri.
Komplikationer og konsekvenser
Forårsaget af virkningen af mellemliggende metabolitter af tyrosin - homogentisinsyre og benzoquinoneddikesyre - opstår konsekvenserne og komplikationerne ved alkaptonuri på grund af aflejring af reaktive pigmenterede polymerer, ødelæggelse af kollagenfibriller og forringelse af bruskens tilstand (med et fald i deres modstandsdygtighed over for mekanisk stress).
Med årene udvikles der i voksenalderen degenerativ gigt og slidgigt i store led (hofte, korsbein og knæ); intervertebrale rum indsnævres (især i lænde- og brysthvirvelsøjlen) – med forkalkning og dannelse af osteofytter; tætheden af vævet i de subkondrale knogleplader falder, og de underliggende knogler kan undergå patologisk ombygning med dannelse af udvækster og deformation. [ 11 ]
Skader på hjerteklapperne (aorta- og mitralklapperne) og koronararterierne kan observeres – med tegn på koronar hjertesygdom, samt dannelse af sten i nyrerne og prostata – på grund af den samme forkalkning. [ 12 ], [ 13 ]
Diagnosticering alkaptonuri
Typisk er diagnosen af medfødte metaboliske lidelser baseret på undersøgelsen af kroppens biologiske væsker.
Baseret på hvilke tests og reaktioner kan alkaptonuri diagnosticeres? Urinprøver er nødvendige for at detektere homogentisinsyre og bestemme dens niveau (normalt – 20-30 mg pr. dag, forhøjet – 3-8 g). En urinprøve undersøges ved gaskromatografi eller massespektrometri ved hjælp af væskekromatografi; en screeningstest for tilstedeværelsen af jernklorid i urinen er mulig. [ 14 ]
Der findes også en metode til hurtig diagnostik – bestemmelse af alkapton i tørrede urinpletter på papir (efter farveintensitet).
Ved afklaring af diagnosen involverer instrumentel diagnostik (radiografi) identifikation af radiologiske tegn på slidgigt og andre ledpatologier hos patienter.
Diagnosen bekræftes ved hjælp af molekylærgenetiske metoder til diagnosticering af arvelige sygdomme, såsom genetisk testning og DNA-sekventering. [ 15 ]
Differential diagnose
Differentialdiagnose omfatter hæmokromatose og akut leversvigt hos den nyfødte, melaninuri, akut intermitterende porfyri, hæmofagocytisk lymfohistiocytose, primær mitokondriel patologi, leddegigt, ankyloserende spondylitis.
Hvem skal kontakte?
Behandling alkaptonuri
Den primære behandling for alkaptonuri er oral administration af store doser (mindst 1000 mg pr. dag) ascorbinsyre. Hos børn øger dette udskillelsen af homogentisinsyre i urinen, og hos voksne reducerer det urinindholdet af dets derivat, benzoquinoneddikesyre, og forsinker dets binding til bindevævsstrukturer i leddene og kollagen. [ 16 ]
Vesteuropæiske klinikker tester lægemidlet Nitisinone (Orfalin), et lægemiddel fra gruppen af metabolitter, der hæmmer den anden fase af tyrosinkatabolismen: omdannelsen af para-hydroxyphenylpyruvat til homogentisinsyre. Brugen af dette farmakologiske middel fører dog til ophobning af tyrosin og kan forårsage alvorlige bivirkninger, herunder uklarhed i hornhinden og fotofobi, næseblod og maveblødning, leversvigt, ændringer i blodet osv. Ikke desto mindre er Nitisinone i USA godkendt af FDA til behandling af type I tyrosinæmi. [17 ], [ 18 ]
Derfor udføres fysioterapibehandling – træningsterapi for at øge muskelstyrken og forbedre ledmobiliteten, balneoterapi og peloidbehandling for at begrænse smerter – ved ledproblemer forårsaget af alkaptonuri.
Selvom tyrosin ikke kun tilføres fra mad, men også produceres i kroppen, anbefales det for patienter med alkaptonuri at følge en proteinfattig kost og begrænse indtaget af fødevarer med højt tyrosinindhold, primært oksekød og svinekød, mejeriprodukter (især oste), bælgfrugter, nødder og frø.
Forebyggelse
Forebyggelse af genmutationer er umulig, men for at forhindre fødslen af børn med høj risiko for medfødte lidelser, findes der medicinsk genetisk rådgivning, som er nødvendig før en planlagt graviditet for de par, hvis familiehistorie inkluderer arvelige sygdomme. [ 19 ]
Vejrudsigt
Dødelig udgang som følge af alkaptonuri er meget sjælden, og døden kan være forårsaget af alvorlige komplikationer, der involverer hjertet og nyrerne. Så den samlede forventede levetid for personer med alkaptonuri er god.
Men livskvaliteten reduceres på grund af intense smerter i leddene eller rygsøjlen med betydelig begrænsning af mobiliteten, ofte progressiv.