Medicinsk ekspert af artiklen

Nye publikationer
Neuroblastom hos børn: årsager, diagnose, behandling
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
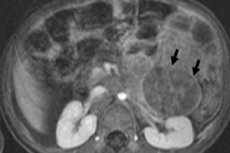
I pædiatrisk onkologi er en af de mest almindelige ekstrakranielle neoplasmer neuroblastom hos børn, som er en embryonal malign tumor i neurale crest-neuroblaster, det vil sige embryonale (umodne) nerveceller i det sympatiske nervesystem.
Epidemiologi
Ifølge statistikker fra International Neuroblastoma Risk Group (INRG) tegner neuroblastom sig for omkring 8% af alle onkologiske sygdomme hos børn på verdensplan og er den tredjestørste prævalens efter leukæmi og hjernetumorer.
Ifølge andre data tegner neuroblastom sig for omkring 28 % af alle kræfttilfælde hos spædbørn. Mere end en tredjedel af tilfældene af neuroblastom diagnosticeres hos børn under et år; den gennemsnitlige diagnosealder er 19-22 måneder. Mere end 90 % af de diagnosticerede tilfælde forekommer hos børn i alderen to til fem år (med en overvægt af drenge); den højeste forekomst observeres i alderen to til tre år, og tilfælde hos børn over fem år tegner sig for mindre end 10 %.
Årsager neuroblastomer
I undersøgelsen af årsagerne til neuroblastom har forskere konkluderet, at denne tumor hos børn opstår på grund af sporadiske genetiske mutationer under embryogenesen eller tidlig postnatal udvikling. Men årsagerne til disse genændringer er ukendte, da der ikke er identificeret nogen indflydelse fra teratogene miljøfaktorer.
Disse tumorer kan forekomme overalt, herunder mediastinum, nakke, mave, binyrer, nyrer, rygsøjle og bækken.
I sjældne tilfælde kan neuroblastom hos spædbørn være forbundet med en arvelig mutation. Især en mutation i genet for membranproteinet CD246 på kromosom 2 - enzymet tyrosinkinase ALK, som sikrer intercellulær kommunikation og spiller en vigtig rolle i nervesystemets funktion; i genet for proteinet PHOX2B (på kromosom 4), som er involveret i modningen af nerveceller.
Neuroblastom kan også være forbundet med neurofibromatose type 1 i barndommen,Beckwith-Wiedemann syndrom og hyperinsulinæmisk hypoglykæmi (nesidioblastose i pankreatitis).
Risikofaktorer
I dag anerkendes arvelighed som risikofaktorer for udvikling af neuroblastom hos børn - tilstedeværelsen af denne tumor i familiens historie, såvel som medfødte anomalier forbundet med genmutationer under intrauterin udvikling. Dette gælder især for tilfælde af udvikling af flere neoplasmer i forskellige organer.
Ingen af de eksogene faktorer, der øger risikoen for denne tumor, er blevet identificeret af forskere.
Patogenese
Mekanismen for udvikling af neuroblastomer skyldes forstyrrelser i differentiering og modning af neurale kamceller – bilaterale cellelinjer, der dannes ved kanterne af neuralrøret fra det ektodermale kimlag i det menneskelige embryo. Disse celler migrerer (bevæger sig) og differentierer til mange typer celler: sensoriske og autonome neuroner, neuroendokrine celler og celler i binyremarven, celler i kraniofacial brusk og knogler samt pigmentceller.
Ved neuroblastom modnes de migrerede neuroblaster ikke, men fortsætter med at vokse og dele sig og danne en tumor. Og patogenesen for dens dannelse er forbundet med følgende genmutationer:
- med duplikering af en del af kromosomsekvensen eller duplikering af segmenter af LMO1-genet på kromosom 11, der koder for RBTN1-proteinet i embryonets neurale kamceller;
- med en ændring i kopitallet af NBPF10-genet på kromosom 1q21.1, der koder for DUF1220-proteinet, som kontrollerer proliferationen af humane neurale stamceller. Disse lidelser fører enten til en duplikering af dette kromosom eller til dets deletion - fraværet af en del af DNA'et;
- med ændringer i tumorsuppressorgenet ATRX (på kromosom Xq21.1);
- med tilstedeværelsen af yderligere kopier (amplifikation) af N-Myc-transkriptionsfaktorgenet på kromosom 2, som koder for en af transkriptionsfaktorerne (DNA-bindende protein), der regulerer aktiviteten af andre gener og kontrollerer proliferationen af precursorceller under dannelsen af proteiner til dannelse af fostervæv og organer. Amplifikation af dette gen omdanner det til et onkogen, hvilket fremkalder en forstyrrelse af cellecyklussen, øget celleproliferation og tumordannelse.
Symptomer neuroblastomer
De første tegn på neuroblastom er uspecifikke og kan omfatte appetitløshed (og vægttab), træthed ved fodring, feber og ledsmerter.
Kliniske symptomer afhænger af den primære tumors placering og tilstedeværelsen af metastaser (som forekommer i 60-73% af tilfældene).
Meget ofte er primært neuroblastom lokaliseret i binyremarven, som har en lignende oprindelse som nerveceller. Hos børn under et år diagnosticeres binyreneuroblastom i 35-40% af tilfældene. Symptomer omfatter mavesmerter, feber, vægttab, knoglesmerter, anæmi eller samtidig Peppers syndrom: diffus leverskade med svær hepatomegali og respiratorisk distresssyndrom.
Retroperitoneal neuroblastom eller retroperitoneal neuroblastom hos børn begynder, efterhånden som det vokser, at presse på blæren eller tarmene, hvilket kan forårsage problemer med vandladning eller afføring, hævelse af benene (hos drenge hæver pungen).
Neuroblastom i mediastinum hos børn (mediastinal neuroblastom) trykker ofte på vena cava superior, og dette kan forårsage hævelse af ansigt, hals, arme og øvre del af brystkassen (hvor huden bliver blåligrød med subkutane knuder). Hoste og hvæsen, vejrtrækningsproblemer (åndenød) eller synkeproblemer (dysfagi) optræder; forstørrede lymfeknuder ses på halsen, over kravebenet og i armhulerne.
Spredning af tumorceller til knoglemarven fører til anæmi, trombocytopeni og leukopeni med tendens til blødning.
Og med metastaser i det periorbitale område opstår der mørke rande eller blå mærker omkring øjnene. En sådan tumor kan også forårsage hovedpine og svimmelhed, exoftalmi (udbuling af øjenæblerne), og på grund af kompression af nerveenderne - hængende øjenlåg (ptosis) og et fald i pupillernes størrelse (miose).
Abdominal neuroblastom eller neuroblastom i bughulen hos børn fører til dannelse af håndgribelige forseglinger i maven, dens udspiling, appetitløshed, forstoppelse og forhøjet blodtryk. En tumor, der trykker på rygmarven eller nerveroden, kan føre til følelsesløshed og svaghed i lemmerne, manglende evne til at stå, kravle eller gå. Hvis knoglerne er påvirket, kan der opstå knoglesmerter.
I tilfælde af tumor i stadium 3-4 i bughulen med lymfeknudebeskadigelse kan tumorceller trænge ind i nyreparenkym, og derefter udvikler der sig et omfattende neuroblastom i nyren hos børn, hvilket fører til forstyrrelse af dens funktioner.
Niveauer
- Stadie 1 neuroblastom er en primær tumor, der er lokaliseret og isoleret til ét område af kroppen; lymfeknuder på begge sider påvirkes ikke.
- Neuroblastom stadie 2. I stadie 2A er den primære tumor begrænset til ét område, men er stor; bilaterale lymfeknuder er ikke involveret. I stadie 2B er lymfeknuderne på den side af kroppen, hvor tumoren er placeret, positive for metastaser.
- Neuroblastom stadie 3: den primære tumor krydser rygmarven eller kroppens midterlinje, ensidige eller bilaterale metastaser findes i lymfeknuderne.
- Neuroblastom stadie 4: Tumoren har spredt sig til fjerne lymfeknuder, knoglemarv, knogler, lever eller andre organer. Stadie 4S bestemmes hos børn under et år med en lokaliseret primær tumor med spredning til hud, lever eller knoglemarv.
Internationalt system til risikoinddeling af neuroblastom (INRGSS)
INRGSS bruger billeddiagnostiske risikofaktorer (IDRF'er), som er faktorer, der ses på billeddiagnostiske tests, og som kan betyde, at en tumor vil være vanskeligere at fjerne.
INRGSS opdeler neuroblastomer i 4 stadier:
- L1: Tumoren har ikke spredt sig fra hvor den startede og er ikke vokset til vitale strukturer. Den er begrænset til én del af kroppen, såsom halsen, brystet eller maven.
- L2: Tumoren har ikke spredt sig (metastaseret) langt fra, hvor den startede (for eksempel kan den være vokset fra venstre side af maven til venstre side af brystet), men den har mindst én IDRF.
- M: Tumoren har metastaseret til en fjern del af kroppen (med undtagelse af tumorer i MS-stadiet).
- MS: Metastatisk sygdom hos børn under 18 måneder, hvor kræften kun har spredt sig til huden, leveren og/eller knoglemarven.
Komplikationer og konsekvenser
Neuroblastom er karakteriseret ved komplikationer og konsekvenser såsom:
- spredning (metastase) til lymfeknuder, knoglemarv, lever, hud og knogler;
- kompression af rygmarven (som kan forårsage smerte og føre til lammelse);
- udvikling af paraneoplastisk syndrom (på grund af virkningen af visse kemikalier udskilt af tumoren, såvel som antigenet disialogangliosid GD2 udtrykt af dens celler), som manifesterer sig ved hurtige ufrivillige øjenbevægelser, nedsat koordination, muskelkramper og diarré;
- tilbagefald efter afslutning af primær behandling (som klinisk praksis viser, har højrisiko-neuroblastomer et tilbagefald i 50% af tilfældene).
Diagnosticering neuroblastomer
Diagnose af mistanke om neuroblastom hos et barn kræver undersøgelse, laboratorietests og billeddiagnostik.
Blod- og urinprøver tages for katekolaminer (norepinephrin og dopamin) og homovanillin- eller vanillylmandelsyrer (dannet under metabolismen af disse hormoner); en blodprøve for neurospecifik enolase, et enzymbundet immunosorbentassay (ELISA) af blodserum og en knoglemarvsanalyse (hvoraf en prøve tages ved aspirationspunktur). En DNA-test udføres for at bestemme mutationer, og en biopsi udføres til cytomorfologisk undersøgelse af tumorvævet.
Efter biopsiprøverne er taget, sendes de til et laboratorium, hvor de undersøges under et mikroskop af en patolog (en læge med særlig træning i at identificere kræftceller). Der udføres også ofte særlige laboratorietests på prøverne for at vise, om tumoren er neuroblastom.
Hvis det er neuroblastom, kan laboratorietests også hjælpe med at bestemme, hvor hurtigt tumoren kan vokse eller sprede sig, samt hvilke behandlinger der kan virke bedst.
Instrumentel diagnostik visualiserer neoplasmen ved hjælp af ultralyd, røntgen, MR eller CT, PET med introduktion af 18F-fluorodeoxyglucose eller MIBG-scanning - scintigrafi med metaiodobenzylguanidin. [ 1 ]
Differential diagnose
Differentialdiagnose omfatter benign ganglioneurom, ganglioneuroblastom, rhabdomyosarkom og nefroblastom.
Hvem skal kontakte?
Behandling neuroblastomer
Ved neuroblastom afhænger behandlingen af patientens risikogruppe (stadie af tumorprocessen), tumorens lokalisering, tumorcellernes genomiske træk og barnets alder. Og den kan omfatte overvågning, kirurgi, kemoterapi, strålebehandling, immunterapi og hæmatopoietisk stamcelletransplantation.
Neoadjuverende eller adjuverende (præ- eller postoperativ) kemoterapi mod neuroblastom hos børn, ligesom al kemoterapi mod kræft, gives i forløb: lægemidlet administreres i flere dage i træk, efterfulgt af en pause, hvor kroppen kan komme sig. Cyklusserne gentages normalt hver tredje til fjerde uge.
Følgende lægemidler (og deres kombinationer) anvendes: Cyclophosphamid, cisplatin eller carboplatin, doxorubicin (adriamycin), vincristin, etoposid.
Almindelige bivirkninger af kemoterapimedicin omfatter hårtab, appetitløshed, træthed, kvalme og opkastning, mundsår, diarré eller forstoppelse. Kemoterapi kan skade knoglemarven og forårsage et fald i blodtallet.
Målrettet immunterapi (rettet mod tumorantigenet GD2) bruger lægemidler fra gruppen af monoklonale antistoffer (anti-GD2 MAb) Dinutuximab (Unituxin) og Naxitamab. De administreres intravenøst ved forlænget infusion i kombination med granulocyt-makrofag-kolonistimulerende faktor (cytokin GM-CSF) og interleukin-2.
Bivirkninger af disse lægemidler omfatter smerter (ofte meget alvorlige), nedsat blodtryk, øget hjertefrekvens, åndenød (med mulig hævelse af luftvejene), øget temperatur, kvalme, opkastning og diarré, ændringer i blodets cellulære og mineralske sammensætning.
For at reducere risikoen for tilbagefald af kræft efter højdosis kemoterapi og stamcelletransplantation behandles børn med højrisiko-neuroblastom med systemiske retinoider, 13-cis-retinsyre (isotretinoin). [ 2 ]
Kirurgisk behandling af neuroblastom – fjernelse af tumor, for eksempel åben adrenalektomi eller laparoskopisk resektion af binyreneuroblastom; lymfektomi (fjernelse af berørte lymfeknuder) osv. [ 3 ]
Ved højrisiko-neuroblastom kan strålebehandling anvendes.[ 4 ]
Forebyggelse
I betragtning af årsagerne til neuroblastom hos børn kan den eneste forebyggende foranstaltning være genetisk rådgivning ved planlægning af graviditet. Men det skal huskes, at denne tumor kun er forbundet med arvelige mutationer i 1-2% af tilfældene.
Vejrudsigt
Infantilt neuroblastom har evnen til spontan regression.
Prognostiske markører
- Højrisikotumorer, såvel som neuroblastom hos børn i alle aldersgrupper og alle stadier (undtagen stadie 4S) – med øget ekspression af N-MYC-genet og amplifikation af N-Myc-onkogenet – har en ugunstig prognose, der påvirker den forventede levealder.
- Tumorceller, der mangler visse dele af kromosom 1 eller 11 (kendt som 1p- eller 11q-deletioner), har en dårligere prognose. En ekstra del af kromosom 17 (17q-forøgelse) er også forbundet med en dårligere prognose.
- Neuroblastomceller med en stor mængde DNA har en bedre prognose, især for børn under 2 år.
- Neuroblastomer, der har flere neurotrofinreceptorer, især nervevækstfaktorreceptoren TrkA, har en bedre prognose.
Overlevelse efter risikogruppe for børneonkologi (COG)
- Lavrisikogruppe: Børn i lavrisikogruppen har en 5-års overlevelsesrate på over 95 %.
- Mellemrisikogruppe: Børn i mellemrisikogruppen har en 5-års overlevelsesrate på 90% til 95%.
- Højrisikogruppe: Børn i højrisikogruppen har en 5-års overlevelsesrate på omkring 50 %.
Omkring 15 % af dødsfaldene i forbindelse med kræft hos børn skyldes neuroblastom. Chancerne for langtidsoverlevelse for denne højrisikomalignitet er ikke mere end 40 %. Den samlede femårsoverlevelsesrate er 67-74 %, 43 % i aldersgruppen et til fire år og mere end 80 % for neuroblastom diagnosticeret i det første leveår.
Использованная литература