Medicinsk ekspert af artiklen

Nye publikationer
Cystisk fibrose
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Cystisk fibrose er en genetisk autosomal recessiv monogen sygdom, der er karakteriseret ved en forstyrrelse af sekretionen af eksokrine kirtler i vitale organer med skade primært på luftvejene og fordøjelsessystemet, et alvorligt forløb og en ugunstig prognose.
 [ 1 ]
[ 1 ]
Epidemiologi
Forekomsten af cystisk fibrose svinger mellem 1:2.500 og 1:4.600 nyfødte. Hvert år fødes der omkring 45.000 personer med cystisk fibrose på verdensplan. Forekomsten af bærere af cystisk fibrose-genet er 3-4%, hvor omkring 275 millioner mennesker på verdensplan er bærere af dette gen, hvoraf omkring 5 millioner bor i Rusland og omkring 12,5 millioner i SNG-landene.
Årsager cystisk fibrose
Cystisk fibrose overføres autosomalt recessivt. Cystisk fibrose-genet er placeret i autosom 7, indeholder 27 exoner og består af 250.000 nukleotidpar.
Et enkelt gen kan have mange mutationer, som hver især er specifik for en bestemt population eller geografisk region. Mere end 520 mutationer er blevet beskrevet, hvoraf den mest almindelige er delta-P-508, dvs. en substitution af aminosyren phenylalanin i position 508.
Patogenese
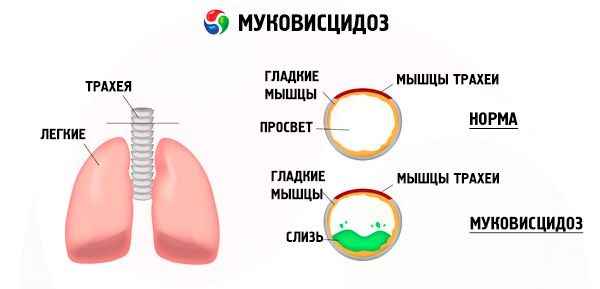
Mutationer i genet for cystisk fibrose forstyrrer strukturen og funktionen af et protein kaldet CFTR (cystisk fibrose transmembranregulator). Dette protein fungerer som en kloridkanal, der er involveret i vand-elektrolytudvekslingen i epitelceller i det bronkopulmonale system, mave-tarmkanalen, bugspytkirtlen, leveren og reproduktionssystemet. Som følge af forstyrrelsen af CFTR-proteinets funktion og struktur akkumuleres kloridioner Cl- inde i cellen. Dette fører til en ændring i det elektriske potentiale i lumen i udskillelseskanalerne, hvilket letter strømmen af store mængder natriumioner (Na + ) fra kanalens lumen ind i cellen og yderligere forbedrer absorptionen af vand fra det pericellulære rum.
Som følge af disse ændringer fortykkes udskillelsen af de fleste eksokrine kirtler, dens evakuering forstyrres, hvilket fører til udtalte sekundære lidelser i organer og systemer, mest udtalt i bronkopulmonale og fordøjelsessystemer.
En kronisk inflammatorisk proces af varierende intensitet udvikler sig i bronkierne, funktionen af det cilierede epitel forstyrres kraftigt, sputumet bliver meget viskøst, tykt, meget vanskeligt at tømme ud, dets stagnation observeres, bronkiolo- og bronkiektasi dannes, som over tid bliver mere almindelige. Disse ændringer fører til en øget hypoxi og dannelsen af kronisk pulmonal hjertesygdom.
Patienter med cystisk fibrose er ekstremt prædisponerede for udvikling af kronisk inflammation i det bronkopulmonale system. Dette skyldes udtalte forstyrrelser i det lokale bronkopulmonale forsvarssystem (reducerede niveauer af IgA, interferon, fagocytisk funktion af alveolære makrofager og leukocytter).
Alveolære makrofager spiller en vigtig rolle i udviklingen af kronisk inflammation i det bronkopulmonale system. De producerer store mængder IL-8, hvilket dramatisk øger neutrofil kemotaksi i bronkialtræet. Neutrofiler akkumuleres i store mængder i bronkierne og udskiller sammen med epitelceller mange proinflammatoriske cytokiner, herunder IL-1, 8, 6, tumornekrosefaktor og leukotriener.
En vigtig rolle i patogenesen af skader på det bronkopulmonale system spilles også af den høje aktivitet af enzymet elastase. Der skelnes mellem eksogen og endogen elastase. Den første produceres af bakteriefloraen (især Pseudomonas aeruginosa), den anden - af neutrofile leukocytter. Elastase ødelægger epitelet og andre strukturelle elementer i bronkierne, hvilket bidrager til yderligere forstyrrelse af mukociliær transport og den hurtige dannelse af bronkiektasi.
Neutrofile leukocytter udskiller også andre proteolytiske enzymer. Alfa-1-antipyrsin og en sekretorisk hæmmer af leukoproteaser modvirker virkningen af proteolytiske enzymer og beskytter derfor det bronkopulmonale system mod deres skadelige indflydelse. Desværre undertrykkes disse beskyttende faktorer hos patienter med cystisk fibrose dog af en betydelig mængde neutrofil protease.
Alle disse omstændigheder bidrager til introduktionen af infektion i det bronkopulmonale system og udviklingen af kronisk purulent bronkitis. Derudover skal det tages i betragtning, at det defekte protein kodet af cystisk fibrose-genet ændrer den funktionelle tilstand af det bronkiale epitel, hvilket favoriserer bakteriers adhæsion til det bronkiale epitel, primært Pseudomonas aeruginosa.
Sammen med patologien i det bronkopulmonale system forårsager cystisk fibrose også alvorlig skade på bugspytkirtlen, maven, tyktarmen og tyndtarmen samt leveren.
Symptomer cystisk fibrose
Cystisk fibrose manifesterer sig med forskellige kliniske symptomer. Hos nyfødte kan sygdommen manifestere sig med meconium ileus. På grund af mangel på eller endda fuldstændig mangel på trypsin bliver meconium meget tæt, viskøst og akkumuleres i ileocækalregionen. Yderligere udvikles tarmobstruktion, som manifesterer sig ved intens opkastning med tilsætning af galde, abdominal distension, manglende meconiumudskillelse, udvikling af peritonitis-symptomer og en hurtig stigning i kliniske manifestationer af alvorligt forgiftningssyndrom. Barnet kan dø i de første levedage, hvis der ikke udføres akut kirurgisk indgreb.
I mindre alvorlige tilfælde er et karakteristisk tegn på cystisk fibrose rigelig, hyppig afføring, fyldig, med en stor mængde fedt og en meget ubehagelig lugt. Hos 1/3 af patienterne observeres endetarmsprolaps.
Efterfølgende oplever patienterne fortsat tarmdysfunktion, malabsorptionssyndrom, alvorlige fysiske udviklingsforstyrrelser og alvorlig hypovitaminose.
I det første eller andet leveår opstår symptomer på skade på det bronkopulmonale system (en mild form af sygdommen), som manifesterer sig ved en hoste, der kan være ekstremt udtalt og ligne hoste med kighoste. Hosten ledsages af cyanose, åndenød og udskillelse af tykt opspyt, først slimet og derefter purulent. Gradvist dannes et klinisk billede af kronisk obstruktiv bronkitis og bronkiektasi, lungeemfysem og respirationssvigt. Børn er ekstremt modtagelige for akutte respiratoriske virus- og bakterieinfektioner, hvilket bidrager til forværring og progression af bronkopulmonal patologi. Udvikling af infektionsafhængig bronkial astma er mulig.
Hos skolebørn kan cystisk fibrose manifestere sig som "tarmkolik". Patienter klager over svære paroxysmale mavesmerter, oppustethed og gentagen opkastning. Ved palpering af maven bestemmes tætte formationer, der er placeret i tyktarmens projektion - fækale masser blandet med tykt, tæt slim. Børn er meget tilbøjelige til at udvikle hypokloræmisk alkalose på grund af overdreven udskillelse af salt med sved i varmt vejr, mens "saltfrost" opstår på barnets hud.
Lidelser i det bronkopulmonale system hos voksne
Skader på det bronkopulmonale system hos patienter med cystisk fibrose (lungeformen af sygdommen) er karakteriseret ved udvikling af kronisk purulent obstruktiv bronkitis, bronkiektasi, kronisk lungebetændelse, lungeemfysem, respirationssvigt og pulmonal hjertesygdom. Nogle patienter udvikler pneumothorax og andre komplikationer af cystisk fibrose: atelektase, lungeabcesser, hæmoptyse, lungeblødning og infektionsafhængig bronkial astma.
Patienter klager over en smertefuld paroxysmal hoste med meget tyktflydende, vanskeligt udskillende mukopurulent sputum, undertiden med en blanding af blod. Derudover er åndenød ekstremt karakteristisk, først under fysisk anstrengelse og derefter i hvile. Åndenød skyldes bronkial obstruktion. Mange patienter klager over kronisk rhinitis forårsaget af polypose og bihulebetændelse. Betydelig svaghed, progressivt fald i ydeevne og hyppige akutte respiratoriske virussygdomme er også karakteristiske. Ved undersøgelse rettes opmærksomheden mod bleg hud, hævelser i ansigtet, cyanose af de synlige slimhinder og alvorlig åndenød. Med udviklingen af dekompenseret pulmonal hjertesygdom opstår ødem i benene. Fortykkelse af fingrenes terminale phalanges i form af trommestikker og negle i form af urglas kan observeres. Brystet antager en tøndeformet form (på grund af udviklingen af lungeemfysem).
Perkussion af lungerne afslører tegn på emfysem - en kasselyd, en skarp begrænsning af mobiliteten af lungekanten og en sænkning af lungernes nedre kant. Auskultation af lungerne afslører hård vejrtrækning med forlænget udånding, spredt tør hvæsen og fugtig, middel og fin boblende hvæsen. Ved svær lungeemfysem er vejrtrækningen kraftigt svækket.
Ekstrapulmonale manifestationer af cystisk fibrose
Ekstrapulmonale manifestationer af cystisk fibrose kan være ret udtalte og forekommer hyppigt.
Skade på bugspytkirtlen
Utilstrækkelig eksokrine funktion i bugspytkirtlen af varierende sværhedsgrad observeres hos 85% af patienter med cystisk fibrose. Ved mindre skader på bugspytkirtlen er der ingen fordøjelsesproblemer og malabsorptionssyndromer, og der er kun laboratoriemanifestationer af eksokrin insufficiens (lave niveauer af trypsin og lipase i blodet og tolvfingertarmsindholdet; ofte svær steatorrhea). Det er kendt, at for at forebygge fordøjelsesproblemer er kun en sekretion af 1 til 2% af den samlede lipase tilstrækkelig. Kun signifikante forstyrrelser i den eksokrine funktion manifesterer sig klinisk.
Under normale forhold producerer acini i bugspytkirtlen en flydende sekretion rig på enzymer. Når sekretionen bevæger sig langs udskillelseskanalen, beriges den med vand og anioner, og den bliver endnu mere flydende. Ved cystisk fibrose modtager bugspytkirtelsekretionen på grund af en forstyrrelse i strukturen og funktionen af transmembranregulatoren (kloridkanalen) ikke en tilstrækkelig mængde væske, den bliver viskøs, og hastigheden af dens bevægelse langs udskillelseskanalen aftager kraftigt. Sekretionens proteiner aflejres på væggene i små udskillelseskanaler, hvilket resulterer i deres obstruktion. Efterhånden som sygdommen skrider frem, udvikles der i sidste ende ødelæggelse og atrofi af acini - kronisk pankreatitis med eksokrin pankreasinsufficiens dannes. Dette afspejles klinisk i udviklingen af fordøjelses- og malabsorptionssyndromer. Bugspytkirtelinsufficiens er hovedårsagen til fedtmalabsorption ved cystisk fibrose, men dette observeres normalt med en betydelig lipasemangel. Forsher og Durie (1991) angiver, at fedt nedbrydes og absorberes med 50-60% i fuldstændig fravær af pankreatisk lipase, hvilket skyldes tilstedeværelsen af mave- og spytlipaser (sublinguale), hvis aktivitet er tæt på den nedre grænse af normen. Sammen med forstyrrelsen af fedtnedbrydning og -absorption sker der en forstyrrelse af proteinnedbrydning og -reabsorption. Omkring 50% af det protein, der indtages med mad, går tabt med afføringen. Kulhydratabsorptionen påvirkes i mindre grad på trods af mangel på α-amylase, men kulhydratmetabolismen kan forstyrres betydeligt.
Skader på bugspytkirtlen udtrykkes i udviklingen af maldigestion og malabsorptionssyndrom med betydeligt vægttab og rigelig fedtholdig afføring.
Udviklingen af maldigestion- og malabsorptionssyndromer fremmes også af alvorlig dysfunktion af tarmkirtlerne, nedsat udskillelse af tarmsaft og et fald i indholdet af tarmensenzymer i den.
Fordøjelsesproblemer og malabsorptionssyndromer kaldes også den intestinale form for cystisk fibrose.
Nedsat endokrin funktion i bugspytkirtlen (diabetes mellitus) observeres hos patienter med cystisk fibrose i de sene stadier af sygdommen (hos 2% af børn og 15% af voksne)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Lever- og galdevejsskader
Hos 13% af patienter med blandede og intestinale former for cystisk fibrose udvikles levercirrose. Det er mest typisk for mutationerne W128X, delta-P508 og X1303K. Biliær levercirrose med portal hypertension påvises hos 5-10% af patienterne. Ifølge Welch, Smith (1995) påvises kliniske, morfologiske, laboratoriemæssige og instrumentelle tegn på leverskade hos 86% af patienter med cystisk fibrose.
Mange patienter med cystisk fibrose udvikler også kronisk kolecystitis, ofte kalklignende.
Dysfunktion af kønskirtlerne
Patienter med cystisk fibrose kan opleve azoospermi, som er årsagen til infertilitet. Nedsat fertilitet er også typisk for kvinder.
Niveauer
Der er tre sværhedsgrader af pulmonal cystisk fibrose.
Den milde form for cystisk fibrose er karakteriseret ved sjældne eksacerbationer (højst én gang om året); i perioder med remission er kliniske manifestationer praktisk talt fraværende, og patienterne er i stand til at arbejde.
Moderat sværhedsgrad - eksacerbationer observeres 2-3 gange om året og varer omkring 2 måneder eller længere. I eksacerbationsfasen er der en intens hoste med vanskeligt at udskille sputum, åndenød selv ved mindre fysisk anstrengelse, subfebril kropstemperatur, generel svaghed, svedtendens. Samtidig er der en forstyrrelse af bugspytkirtlens eksokrine funktion. I remissionsfasen er arbejdsevnen ikke fuldt genoprettet, åndenød under fysisk anstrengelse forbliver.
Et alvorligt forløb er karakteriseret ved meget hyppige forværringer af sygdommen. Remissioner er praktisk talt fraværende. I det kliniske billede træder alvorlig respirationssvigt i forgrunden, symptomer på kronisk pulmonal hjertesygdom, ofte dekompenseret, med typisk hæmoptyse. Der observeres betydeligt vægttab, patienterne er fuldstændig invalide. Som regel ledsages alvorlig bronkopulmonal patologi af en skarpt udtalt dysfunktion af bugspytkirtlen.
Forms
- Bronkopulmonale læsioner
- Gentagen og tilbagevendende lungebetændelse med et langvarigt forløb.
- Abscesserende lungebetændelse, især hos spædbørn.
- Kronisk lungebetændelse, især bilateral.
- Bronkial astma, der er refraktær overfor traditionel behandling.
- Tilbagevendende bronkitis, bronkiolitis, især ved Pseudomonas aeruginosa-kultur.
- Ændringer i mave-tarmkanalen
- Meconium ileus og dens ækvivalenter.
- Syndrom med nedsat intestinal absorption af ukendt oprindelse.
- Obstruktiv gulsot hos nyfødte med et langvarigt forløb.
- Levercirrose.
- Diabetes mellitus.
- Gastroøsofageal refluks.
- Kolelithiasis.
- Rektal prolaps.
- Ændringer i andre organer og systemer
- Vækst- og udviklingsforstyrrelser.
- Forsinket seksuel udvikling.
- Mandlig infertilitet.
- Næsepolypper.
- Søskende fra familier med cystisk fibrose.
[ 24 ]
Komplikationer og konsekvenser
Komplikationer fra mave-tarmkanalen omfatter:
- Diabetes mellitus udvikles hos 8-12% af patienter over 25 år.
- Fibroserende kolonopati.
- Meconium ileus i den nyfødte periode (hos 12% af nyfødte med cystisk fibrose), distal intestinal obstruktionssyndrom, rektal prolaps, mavesår og gastroøsofageal reflukssygdom.
Leverkomplikationer:
- Fedtleversygdom (hos 30-60% af patienterne)
- Fokal biliær cirrose, multinodulær biliær cirrose og associeret portal hypertension.
Portal hypertension fører undertiden til død på grund af spiserørsvarices.
Forekomsten af kolecystitis og galdesten er højere hos patienter med cystisk fibrose end hos andre individer.
Forsinket pubertet og nedsat fertilitet og andre komplikationer. De fleste mænd har azoospermi og underudvikling af sædlederen.
Diagnosticering cystisk fibrose
Generel blodprøve - typisk anæmi af varierende sværhedsgrad, normalt normo- eller hypokrom. Anæmi har en polyfaktoriel genese (reduceret absorption af jern og vitamin B12 i tarmen på grund af udvikling af malabsorptionssyndrom). Leukopeni er mulig, med udvikling af purulent bronkitis og lungebetændelse - leukocytose, øget ESR.
Generel urinanalyse - ingen signifikante ændringer, i sjældne tilfælde observeres let proteinuri.
Koprologisk undersøgelse - steatorrhea og kreatorrhea observeres. Becker (1987) anbefaler måling af chymotrypsin og fedtsyrer i afføring. Før bestemmelse af chymotrypsin i afføring er det nødvendigt at stoppe med at tage fordøjelsesenzymer mindst 3 dage før undersøgelsen. Ved cystisk fibrose reduceres mængden af chymotrypsin i afføring, og mængden af fedtsyrer øges (normal udskillelse af fedtsyrer er mindre end 20 mmol/dag). Det er nødvendigt at tage højde for, at øget udskillelse af fedtsyrer med afføring også observeres hos:
- mangel på konjugerede fedtsyrer i tyndtarmen på grund af leversvigt, obstruktion af galdegangene, betydelig bakteriel kolonisering af tyndtarmen (i dette tilfælde forekommer intensiv hydrolyse af galdesyrer);
- ileitis;
- cøliaki (med udvikling af malabsorptionssyndrom);
- enteritis;
- intestinale lymfomer;
- Whipples sygdom;
- fødevareallergier;
- accelereret transit af fødevaremasser i diarré af forskellig oprindelse, carcinoid syndrom, thyrotoksikose.
Biokemisk blodprøve - nedsat total protein- og albuminniveau, forhøjede niveauer af alfa2- og gammaglobuliner, bilirubin og aminotransferaser (i tilfælde af leverskade), nedsat aktivitet af amylase, lipase, trypsin samt jern- og calciumniveauer (i tilfælde af udvikling af maldigestionssyndrom, malabsorption).
Sputumanalyse - tilstedeværelsen af et stort antal neutrofile leukocytter og mikroorganismer (under sputumbakterioskopi).
En undersøgelse af tyndtarmens absorptionsfunktion og bugspytkirtlens eksokrine funktion afslører betydelige forstyrrelser.
Røntgenundersøgelse af lungerne - afslører forandringer, hvis sværhedsgrad afhænger af sygdommens sværhedsgrad og fase. De mest karakteristiske forandringer er:
- øget pulmonal mønsterdannelse på grund af peribronkiale interstitielle ændringer;
- udvidelse af lungernes rødder;
- billede af lobulær, subsegmental eller endda segmental atelektase i lungerne;
- øget gennemsigtighed i lungefelterne, primært i de øvre sektioner, lav position og utilstrækkelig mobilitet af membranen, udvidelse af det retrosternale rum (manifestation af lungeemfysem);
- segmental eller polysegmental infiltration af lungevæv (i udviklingen af lungebetændelse).
Bronkografi afslører ændringer forårsaget af obstruktion af bronkierne med viskøs sputum (fragmentering af bronkiernes fyldning med kontrastmiddel, ujævne konturer, fænomenet bronkial ruptur, et signifikant fald i antallet af laterale grene) samt bronkoekgaser (cylindriske, blandede), lokaliseret hovedsageligt i de nedre dele af lungerne.
Bronkoskopi afslører diffus purulent bronkitis med rigeligt tykt, viskøst sputum og fibrinøse film.
Spirometri - allerede i sygdommens tidlige stadier afslører respirationssvigt af den obstruktive type (nedsat FVC, FEV1, Tiffno-indeks), restriktiv (nedsat FVC) eller oftest obstruktiv-restriktiv (nedsat FVC, FVC, FEV1, Tiffno-indeks).
Gibson og Cooks svedetest (svedelektrolyttetest) involverer stimulering af sved ved hjælp af pilocarpinelektroforese med efterfølgende bestemmelse af klorider i sveden. Doerehuk (1987) beskriver testen som følger. Pilocarpinelektroforese udføres på underarmen, den elektriske strøm er 3 mA. Efter rensning af huden med destilleret vand opsamles sveden ved hjælp af filterpapir placeret på det stimulerede område, dækket med gaze for at forhindre fordampning fra det. Efter 30-60 minutter fjernes filterpapiret og elueres i destilleret vand. Mængden af opsamlet sved måles. For at opnå pålidelige resultater er det nødvendigt at opsamle mindst 50 mg (helst 100 mg) sved.
Hvis kloridkoncentrationen er mere end 60 mmol/l, anses diagnosen cystisk fibrose for sandsynlig; hvis kloridkoncentrationen er mere end 100 mmol/l, er den pålidelig; i dette tilfælde bør forskellen i koncentrationen af klor og natrium ikke overstige 8-10 mmol/l. Hadson (1983) anbefaler, at hvis natrium- og kloridindholdet i sved er grænsetilfælde, bør der udføres en prednisolontest (5 mg oralt i 2 dage, efterfulgt af bestemmelse af elektrolytter i sved). Hos personer, der ikke lider af cystisk fibrose, falder natriumniveauet i sved til den nedre normalgrænse; ved cystisk fibrose ændrer det sig ikke. En svedtest anbefales til alle børn med kronisk hoste.
Analyse af blodpletter eller DNA-prøver for større mutationer i cystisk fibrose-genet er den mest følsomme og specifikke diagnostiske test. Denne metode er dog kun egnet til lande, hvor delta-P508-mutationsraten er højere end 80 %. Derudover er teknikken meget dyr og teknisk kompleks.
Prænatal diagnose af cystisk fibrose udføres ved at bestemme isoenzymerne af alkalisk fosfatase i fostervand. Denne metode er mulig fra 18-20 uger af graviditeten.
De vigtigste kriterier for diagnosticering af cystisk fibrose er følgende:
- indikationer i anamnesen af forsinket fysisk udvikling i barndommen, tilbagevendende kroniske luftvejssygdomme, dyspeptiske lidelser og diarré, tilstedeværelsen af cystisk fibrose hos nære slægtninge;
- kronisk obstruktiv bronkitis, ofte tilbagevendende, med udvikling af bronkiektasi og lungeemfysem, ofte tilbagevendende lungebetændelse;
- kronisk tilbagevendende pankreatitis med et markant fald i eksokrin funktion, malabsorptionssyndrom;
- øget klorindhold i patientens sved;
- infertilitet med bevaret seksuel funktion.
En vellykket diagnose og differentialdiagnose af cystisk fibrose fremmes ved at identificere risikogrupper.
Screeningsprogram for cystisk fibrose
- Generel analyse af blod, urin, sputum.
- Bakteriologisk analyse af sputum.
- Koprologisk analyse.
- Biokemisk blodprøve: bestemmelse af totalt protein og proteinfraktioner, glukose, bilirubin, aminotransferaser, alkalisk fosfatase, gamma-glutamyltranspeptidaser, kalium, calcium, jern, lipase, amylase, trypsin.
- Undersøgelse af bugspytkirtlens eksokrine funktion og tarmens absorberende funktion.
- Fluoroskopi og radiografi af lungerne, CT-scanning af lungerne.
- EKG.
- Ekkokardiografi.
- Bronkoskopi og bronkografi.
- Spirometri.
- Svedtest.
- Konsultation med en genetiker.
- Analyse af blodpletter eller DNA-prøver for større mutationer i cystisk fibrose-genet.
Hvad skal man undersøge?
Hvordan man undersøger?
Hvilke tests er nødvendige?
Hvem skal kontakte?
Behandling cystisk fibrose
Typen og sværhedsgraden af symptomer på cystisk fibrose kan variere meget, så der er ingen typisk behandlingsplan; den er individualiseret til hver enkelt person.
Terapien består af følgende terapeutiske foranstaltninger:
- Åndedrætsøvelser og postural dræning hjælper med at slippe af med tykt slim, der ophobes i lungerne. Nogle teknikker til at rense luftvejene kræver hjælp fra familiemedlemmer, venner eller en lungelæge. Mange mennesker bruger en oppustelig brystvest, der vibrerer med en høj frekvens.

- Inhalationslægemidler, der har bronkodilatoriske, drænende (mukolytiske) og antibakterielle virkninger (for eksempel fluorquinoloner).
- Præparater indeholdende bugspytkirtelenzymer til forbedring af fordøjelsen. Disse præparater tages under måltider.
- Multivitaminer (inklusive fedtopløselige vitaminer).
I 2015 godkendte FDA et andet lægemiddel til behandling af cystisk fibrose, der er rettet mod et defekt protein kendt som CFTR. Det første lægemiddel, en såkaldt CFTR-modulator, blev godkendt i 2012. CFTR-modulatorer forventes at forlænge livet for nogle mennesker med cystisk fibrose med årtier.
Kirurgi kan være nødvendig for at behandle følgende respiratoriske komplikationer:
- Pneumothorax, massiv tilbagevendende eller vedvarende hæmoptyse, næsepolypper, vedvarende og kronisk bihulebetændelse.
- Meconium ileus, invagination, rektal prolaps.
Lungetransplantation udføres i sygdommens terminale stadium.
Vejrudsigt
Den gennemsnitlige overlevelsesalder for patienter med cystisk fibrose varierer fra 35 til 40 år. Den gennemsnitlige overlevelsesalder er højere for mænd end for kvinder.
Med moderne behandlingsstrategier når 80% af patienterne voksenalderen. Cystisk fibrose begrænser dog patientens funktionelle evner betydeligt. Der findes stadig ingen kur mod denne sygdom.