Medicinsk ekspert af artiklen

Nye publikationer
Achondroplasi
Sidst revideret: 12.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Der er mange sjældne medfødte sygdomme, og en af dem er en krænkelse af knoglevækst - achondroplasi, hvilket fører til alvorlig uforholdsmæssig kort statur.
I afsnittet om udviklingsanomalier i ICD-10 er koden for denne type arvelig osteokondral dysplasi med vækstdefekter i rørknogler og rygsøjle Q77.4 [ 1 ]
Epidemiologi
Hvad angår forekomsten af achondroplasi, er statistiske data fra forskellige undersøgelser tvetydige. Nogle hævder, at denne anomali forekommer hos én nyfødt ud af 10 tusind, andre - hos én ud af 26-28 tusind, og atter andre - 4-15 tilfælde ud af 100 tusind. [ 2 ]
Der er også information om, at når faderen er over 50 år gammel, er forekomsten af achondroplasi hos børn ét tilfælde pr. 1875 nyfødte.
Årsager achondroplasi
Årsagen til achondroplasi er en krænkelse af osteogenesen, især en af typerne af intrauterin ossifikation af diafyserne i skeletets rørformede knogler - endokondral ossifikation, hvor brusk omdannes til knoglevæv. For flere detaljer, se - Knogleudvikling og vækst
Forstyrrelse af ossifikationen af lange knogler, dvs. føtal achondroplasi, opstår på grund af mutationer i membrantyrosinkinase-genet - fibroblastvækstfaktorreceptor 3 (FGFR3 på kromosom 4p16.3), hvilket påvirker cellevækst og -differentiering. Tilstedeværelsen af FGFR3-mutationer er forbundet med genetisk ustabilitet og ændringer i antallet af kromosomer (aneuploidi).
Akondroplasi overføres til et barn som et autosomalt dominant træk, det vil sige, at barnet modtager én kopi af det mutante gen (som er dominant) og ét normalt gen på et par ikke-kønsrelaterede (autosomale) kromosomer. Således er arvetypen af denne defekt autosomal dominant, og anomalien kan manifestere sig hos 50% af afkommet, når en kombination af alleler af dette gen (genotype) krydses.
Derudover kan mutationer være sporadiske, og som praksis viser, fødes børn med achondroplasi i 80% af tilfældene af forældre med normal højde.
Risikofaktorer
De vigtigste risikofaktorer for fødslen af børn med achondroplasi er arvelige. Hvis en af forældrene har denne defekt, er sandsynligheden for at få et sygt barn estimeret til 50%; hvis begge forældre har denne anomali, er den også 50%, men med en 25% risiko for homozygot achondroplasi, hvilket fører til død før fødslen eller i den tidlige barndom.
Med faderens alder (tættere på 40 år og ældre) øges risikoen for en ny mutation (de novo-mutation) af FGFR3-genet.
Patogenese
Eksperter forklarer patogenesen af achondroplasi og understreger vigtigheden af transmembranproteinet tyrosinproteinkinase (kodet af FGFR3-genet) i reguleringen af deling, differentiering og apoptose af celler i bruskvævet i vækstpladerne - chondrocytter, samt den normale udvikling af skelettet - osteogenese og mineralisering af knoglevæv.
Under embryonal udvikling, i nærvær af en genmutation, bliver receptorerne for fibroblastvækstfaktor 3 mere aktive. Forøgelsen af deres funktioner forstyrrer transmissionen af cellulære signaler og interaktionen mellem den ekstracellulære del af dette protein og polypeptidfibroblastvækstfaktorer (FGF). Som følge heraf opstår der en fejl: bruskcellernes proliferationsstadium bliver kortere, og deres differentiering begynder tidligere end forventet. Alt dette fører til forkert dannelse og fusion af kraniets knogler og skeletdysplasi - et fald i lange knogler, som ledsages af udtalt lav statur eller dværgvækst.
Og to tredjedele af tilfældene af dværgvækst er forbundet med achondroplasi.
Symptomer achondroplasi
Unormal knoglevækst forårsager kliniske symptomer på achondroplasi såsom:
- udtalt lav statur (uforholdsmæssig dværgvækst) med en gennemsnitlig voksenhøjde på 123-134 cm;
- forkortelse af de proximale dele af under- og overekstremiteterne med en relativt normal torsostørrelse;
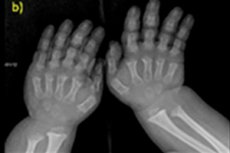
- forkortede fingre og tæer;
- forstørret hoved (makro- eller megalocephali); [ 3 ]
- specifikke ansigtstræk i form af en fremspringende pande og hypoplasi af den midterste del af ansigtet - en nedsænket næserygg.
- smal kraniocervikal overgang. Nogle spædbørn med achondroplasi dør i løbet af deres første leveår af komplikationer relateret til den kraniocervikale overgang; befolkningsstudier tyder på, at denne øgede risiko for død kan være så høj som 7,5 % uden evaluering og intervention.[ 4 ]
- Mellemøredysfunktion er ofte et problem [ 5 ], og hvis den ikke behandles korrekt, kan det føre til konduktivt høretab, der er alvorligt nok til at forstyrre taleudviklingen. Mere end halvdelen af børnene vil have brug for en trykudligningsslange. [ 6 ] Samlet set har omkring 40 % af personer med achondroplasi funktionelt signifikant høretab. Udviklingen af ekspressivt sprog er også ofte forsinket, selvom styrken af sammenhængen mellem høretab og ekspressive sprogproblemer er tvivlsom.
- Bøjning af skinnebenet er meget almindeligt hos personer med achondroplasi. Over 90 % af ubehandlede voksne har en eller anden grad af bøjning.[ 7 ] "Bøjning" er faktisk en kompleks deformitet, der skyldes en kombination af lateral hældning, intern torsion af skinnebenet og dynamisk ustabilitet i knæet.[ 8 ]
Spædbørn med achondroplasi er karakteriseret ved muskulær hypotoni, hvilket gør, at de begynder at lære bevægelsesfærdigheder og gå senere. Intelligens og kognitive evner påvirkes ikke af denne udviklingsdefekt. [ 9 ], [ 10 ]
Konsekvenser og komplikationer
Denne type arvelig osteokondral dysplasi er karakteriseret ved følgende komplikationer og konsekvenser:
- tilbagevendende øreinfektioner;
- obstruktiv søvnapnø;
- hydrocephalus;
- malokklusion og skæve tænder:
- deformation af benene (varus eller valgus) med en ændring i gangart;
- hypertrofieret lordose i lændehvirvelsøjlen eller dens krumning (thoracolumbar kyfose eller lumbal skoliose) - med rygsmerter ved gang;
- ledsmerter (på grund af forkert knogleplacering eller kompression af nerverødder);
- Spinalstenose og rygmarvskompression; Den mest almindelige medicinske klage i voksenalderen er symptomatisk spinalstenose, der involverer L1-L4. Symptomerne spænder fra intermitterende, reversibel claudicatio induceret af træning til alvorlig, irreversibel bendysfunktion og urinretention.[ 11 ] Claudicatio og stenose kan forårsage både sensoriske (følelsesløshed, smerte, tyngde) og motoriske symptomer (svaghed, snublen, begrænset gangudholdenhed). Vaskulær claudicatio skyldes hævelse af blodkar efter stående og gang og er fuldstændig reversibel ved hvile. Spinalstenose er den faktiske læsion af rygmarven eller nerveroden forårsaget af den stenotiske knogle i rygmarvskanalen, og symptomerne er irreversible. Symptomer lokaliseret til et bestemt dermatom kan skyldes stenose af specifikke nerverodsforamina.
- en reduktion af brystvæggen med begrænset lungevækst og nedsat lungefunktion (alvorlig åndenød). I spædbarnsalderen har en lille gruppe mennesker med achondroplasi restriktive lungeproblemer. Små bryster og øget brystcompliance resulterer i kombination i reduceret lungekapacitet og restriktiv lungesygdom [ 12 ]
Andre ortopædiske problemer
- Ledsvaghed. De fleste led er hypermobile i barndommen. Generelt har dette ringe effekt, bortset fra knæinstabilitet hos nogle mennesker.
- Discoid lateral menisk: Denne nyligt identificerede strukturelle abnormalitet kan føre til kroniske knæsmerter hos nogle mennesker.[ 13 ]
- Gigt: Konstitutiv aktivering af FGFR-3, som ved achondroplasi, kan beskytte mod udvikling af gigt.[ 14 ]
- Acanthosis nigricans ses hos cirka 10% af personer med achondroplasi.[ 15 ] I denne population afspejler det ikke hyperinsulinæmi eller malignitet.
Homozygot akondroplasi forårsaget af bialleliske patogene varianter ved nukleotid 1138 af FGFR3 er en alvorlig lidelse med radiologiske fund, der er kvalitativt forskellige fra dem, der ses ved akondroplasi. Tidlig død skyldes respirationssvigt på grund af en lille brystvæg og neurologiske defekter på grund af cervicomedullær stenose [Hall 1988].
Diagnosticering achondroplasi
Hos de fleste patienter stilles diagnosen achondroplasi baseret på karakteristiske kliniske tegn og radiografiske fund. Hos spædbørn eller i fravær af visse symptomer anvendes genetisk testning, såsom karyotypeanalyse, til at stille en endelig diagnose.[16 ]
Ved prænatal diagnostik ved hjælp af den molekylærgenetiske metode kan der udføres analyser af fostervand eller en chorionvillusprøve.
Tegn på achondroplasi på ultralyd af fosteret - forkortelse af lemmerne og typiske ansigtstræk - visualiseres efter 22 ugers graviditet.
Instrumentel diagnostik omfatter også røntgen af skelettet eller ultralyd af knoglerne. Og røntgen bekræfter diagnosen baseret på data som et stort kranium med en smal occipital foramen og en relativt lille base; korte rørformede knogler og forkortede ribben; korte og flade hvirvellegemer; indsnævret rygmarvskanal, reduceret størrelse af iliacvingerne.
Differential diagnose
Differentialdiagnostik ved hypofysedværgvækst, medfødt spondyloepifyseal og diastrofisk dysplasi, hypokondroplasi, Shereshevsky-Turner og Noonan syndromer, pseudoakondroplasi er nødvendig. Forskellen mellem pseudoakondroplasi og akondroplasi er således, at hos patienter med dværgvækst ved pseudoakondroplasi er hovedstørrelsen og ansigtstrækkene normale.
Hvem skal kontakte?
Behandling achondroplasi
Anbefalinger til pleje af børn med achondroplasi er blevet udarbejdet af American Academy of Pediatrics Committee on Genetics. Disse anbefalinger er ment som vejledning og erstatter ikke individuel beslutningstagning. En nylig gennemgang [Pauli & Botto 2020] indeholder også retningslinjer. Der findes specialklinikker, der specialiserer sig i behandling af skeletdysplasi; deres anbefalinger kan afvige en smule fra disse generelle anbefalinger.
Anbefalingerne omfatter (men er ikke begrænset til) følgende.
Hydrocephalus. Hvis der opstår tegn eller symptomer på forhøjet intrakranielt tryk (f.eks. accelereret hovedvækst, vedvarende udbulende fontanel, mærkbar forstørrelse af overfladiske vener i ansigtet, irritabilitet, opkastning, synsforandringer, hovedpine), er henvisning til en neurokirurg nødvendig.
Den formodede ætiologi for hydrocephalus ved achondroplasi er øget intrakranielt venetryk på grund af stenose af jugularforamina. Derfor har standardbehandlingen været ventrikuloperitoneal shunting. Endoskopisk tredje ventrikulostomi kan dog være gavnlig hos nogle individer,[ 17 ] hvilket antyder, at andre mekanismer, såsom obstruktion af fjerde ventrikeludløb på grund af kraniocervikal stenose, kan være involveret.[ 18 ]
Kraniocervikal overgangsstenose. Bedste prædiktorer for behov for suboccipital dekompression:
- Hyperrefleksi eller klonus i underekstremiteterne
- Central hypopnø ved polysomnografi
- Reduktion i foramen magnum-størrelse bestemt ved computertomografi af den kraniocervikale overgang og sammenlignet med normer for børn med achondroplasi.[ 19 ]
- Tegn på rygmarvskompression og/eller T2-vægtede signalabnormaliteter er for nylig blevet foreslået som en anden faktor at overveje, når man beslutter sig for at operere.
Hvis der er tydelige tegn på symptomatisk kompression, bør der straks henvises til en pædiatrisk neurokirurg med henblik på dekompressionskirurgi. [ 20 ]
Behandling af obstruktiv søvnapnø kan omfatte:
- Adenotonsillektomi
- Positivt luftvejstryk
- Trakeostomi i ekstreme tilfælde
- Vægttab
Disse interventioner kan resultere i forbedring af søvnforstyrrelser og en vis forbedring af neurologisk funktion.[ 21 ]
I sjældne tilfælde, hvor obstruktionen er alvorlig nok til at kræve trakeostomi, er midface-fremføringskirurgi blevet anvendt til at lindre obstruktion af de øvre luftveje.[ 22 ]
Dysfunktion i mellemøret. Hyppige mellemøreinfektioner, vedvarende væske i mellemøret og efterfølgende høretab bør behandles aggressivt efter behov. Langtidsbehandling med sonde anbefales, da de ofte er nødvendige indtil syv-otteårsalderen.[ 23 ]
Når der opstår problemer i alle aldre, anbefales det at anvende passende behandlingsmetoder.
Kort statur. Adskillige studier har evalueret væksthormonbehandling (GH) som en mulig behandling for achondroplasi ved kort statur.[ 24 ]
Samlet set viser disse og andre serier en indledende acceleration af væksten, men effekten aftager over tid.
I gennemsnit kan man forvente en stigning i voksenhøjden på kun omkring 3 cm.
For nogle er forlængelse af lemmerne fortsat en mulighed ved hjælp af forskellige teknikker. Der kan opnås højdeforøgelser på op til 30-35 cm. [ 25 ] Komplikationer er almindelige og kan være alvorlige.
Mens nogle går ind for at udføre disse procedurer så tidligt som seks til otte år, går mange børnelæger, kliniske genetikere og etikere ind for at udsætte en sådan operation, indtil en ung person er i stand til at deltage i at træffe en informeret beslutning.
I hvert fald i Nordamerika vælger kun en lille andel af de berørte personer at gennemgå avanceret lemforlængelse. Little People of America Medical Advisory Board har udsendt en erklæring vedrørende brugen af avanceret lemforlængelse.
Fedme: Forebyggende foranstaltninger til fedme bør begynde i den tidlige barndom. Standardbehandlinger for fedme bør være effektive hos personer med achondroplasi, selvom kaloriebehovet er lavere. [ 26 ]
Standardvægt- og vægt-i-højde-diagrammer specifikke for achondroplasi bør anvendes til at spore fremskridt. Det er vigtigt at bemærke, at disse kurver ikke er perfekte vægt-i-højde-kurver; de blev udledt af tusindvis af datapunkter fra personer med achondroplasi.
BMI-standarder (Body Mass Index) blev udviklet til børn i alderen 16 år og derunder. [ 27 ] BMI er ikke standardiseret for voksne med achondroplasi; sammenligninger med BMI-kurver for gennemsnitshøjde vil give misvisende resultater. [ 28 ]
Varusdeformitet. Årlig ortopædisk opfølgning af enten en læge med kendskab til achondroplasi eller en ortopædkirurg anbefales. Kriterier for kirurgisk indgreb er blevet offentliggjort.[ 29 ]
Tilstedeværelsen af en progressiv symptomatisk kurve kræver henvisning til en ortopædlæge. Asymptomatisk varusdeformitet i sig selv kræver normalt ikke kirurgisk korrektion. Forskellige interventioner kan vælges (f.eks. guidet vækst ved hjælp af otte plader, valgusosteotomi og derotationsosteotomi). Der findes ingen kontrollerede studier, der sammenligner resultaterne af behandlingsmulighederne.
Kyfose. Spædbørn med achondroplasi udvikler ofte en fleksibel kyfose. Der findes en protokol, der kan hjælpe med at forhindre udviklingen af en fast vinkelkyfose, hvilket inkluderer at undgå fleksible klapvogne, gynger og bæreseler. Råd mod ustøttet sidden; læg altid modtryk på ryggen, når du holder babyen.
- Kyfose forbedres betydeligt eller forsvinder hos de fleste børn efter at have indtaget en ortograd stilling og begyndt at gå. [ 30 ]
- Hos børn, der ikke spontant aftager efter øget kropsstyrke og begyndt at gå, er afstivning normalt tilstrækkeligt til at forhindre persisterende thorakolumbal kyfose.[ 31 ]
- Hvis svær kyfose fortsætter, kan rygkirurgi være nødvendig for at forhindre neurologiske komplikationer.[ 32 ]
Spinalstenose: Hvis der opstår alvorlige tegn og/eller symptomer på spinalstenose, er det nødvendigt med en hurtig henvisning til en kirurgisk specialist.
Udvidet og bred laminektomi anbefales normalt. Relevansen af proceduren afhænger af niveauet (f.eks. thorax eller lumbal) og graden af stenose. Patienterne havde bedre resultater og forbedret funktion, jo hurtigere de blev opereret efter symptomernes debut [ 33 ].
Vaccinationer: Intet ved akondroplasi udelukker alle rutinemæssige vaccinationer. I betragtning af den øgede respiratoriske risiko er DTaP-, pneumokok- og influenzavacciner særligt vigtige.
Tilpasningsbehov: På grund af lav statur kan det være nødvendigt at tilpasse omgivelserne. I skolen kan dette omfatte taburetter, sænkede lyskontakter, toiletter i passende højde eller andre tilgængelighedsmidler, lavere borde og fodstøtter foran stole. Alle børn bør kunne forlade bygningen selvstændigt i tilfælde af en nødsituation. Små hænder og svage sener kan gøre finmotorikken vanskelig. Passende tilpasninger omfatter brug af et mindre tastatur, vægtede penne og glattere skriveflader. De fleste børn bør have en IEP eller en 504-plan.
Pedalforlængere er næsten altid nødvendige til cykling. Modifikationer af arbejdsstationer såsom lavere borde, mindre tastaturer, trin og adgang til toilettet kan også være nødvendige.
Socialisering: På grund af den meget mærkbare korte statur forbundet med achondroplasi, kan berørte personer og deres familier have svært ved at socialisere og tilpasse sig skolen.
Støttegrupper som Little People of America, Inc (LPA) kan hjælpe familier med at håndtere disse problemer gennem støtte fra ligemænd, personligt eksempel og sociale bevidsthedsprogrammer.
Information om beskæftigelse, uddannelse, handicaprettigheder, adoption af lave børn, medicinske problemer, passende tøj, hjælpemidler og forældremyndighed er tilgængelig via et nationalt nyhedsbrev, seminarer og workshops.
Der findes ingen medicin eller ikke-medicinsk behandling, der kan helbrede denne medfødte defekt.
Fysioterapi anvendes oftest; behandling kan også være nødvendig for hydrocephalus (ved shunt eller endoskopisk ventrikulostomi), fedme, [ 34 ] apnø, [ 35 ] mellemøreinfektion eller spinalstenose.
På nogle klinikker foretager de kirurgisk behandling, når barnet er fem til syv år: forlængelse af knoglerne i skinneben, lår og endda skulderben eller korrigering af deformiteten - ved hjælp af operationer og specielle ortopædiske apparater - i tre til fire faser, der hver varer op til 6-12 måneder.
Terapi under undersøgelse
Administration af en C-type natriuretisk peptidanalog er i øjeblikket under kliniske forsøg. De indledende resultater har vist, at det er veltolereret og resulterer i en stigning i væksthastigheden fra baseline hos børn med achondroplasi ( forsøgssted ). [ 36 ] Konjugeret C-type natriuretisk peptid er også i øjeblikket under kliniske forsøg. [ 37 ] Andre overvejelser omfatter tyrosinkinasehæmning [ 38 ], meclizin [ 39 ] og en opløselig rekombinant human FGFR3-lokkekur. [ 40 ]
Søg på clinicaltrials.gov i USA og EU's register over kliniske forsøg i Europa for information om kliniske forsøg med en bred vifte af sygdomme og tilstande.
Forebyggelse
Den eneste forebyggende foranstaltning er prænatal diagnostik af medfødte sygdomme. [ 41 ], [ 42 ]
Vejrudsigt
Hvor længe lever personer med achondroplasi? Omkring 10 år kortere end den gennemsnitlige forventede levetid.
Da patologiske forandringer i knoglevæv og led fører til begrænsninger i egenomsorg og mobilitet, får børn med denne diagnose status som handicappede. På lang sigt har de fleste patienter en normal prognose, men med alderen er der en øget risiko for hjertesygdomme. [ 43 ]