Medicinsk ekspert af artiklen

Nye publikationer
Arvelig nefritis (Alport syndrom) hos børn
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Arvelig nefritis (Alport syndrom) - genetisk bestemt ikke-immunt arvet glomerulopati udviser hæmaturi (undertiden proteinuri), progressivt fald i nyrefunktion i kronisk nyresvigt udvikling er ofte forbundet med sensorineural døvhed og synshandicappede.
For første gang blev sygdommen beskrevet i 1902 af LGGuthrie, der observerede en familie i flere generationer, hvoraf hæmaturi blev observeret. I 1915 beskrev medlemmer af samme AFHurst-familie udviklingen af uremi. I 1927 identificerede A Alport først døvhed hos flere pårørende med hæmaturi. I 50'erne i forrige århundrede blev øjenskader beskrevet i en sådan sygdom. I 1972 undersøgte Hinglais et al. Hos patienter med arvelig hæmaturi morfologisk undersøgelse af renalvæv. Afslørede ujævn ekspansion og delaminering af glomerulære basale membraner. I 1985 blev det genetiske grundlag for arvelig nefritis - en mutation i genet af type IV-collagen (Fiengold et al., 1985) identificeret.
Undersøgelse af sygdommens genetiske karakter gjorde det muligt at konkludere, at forskelle i fænotypiske manifestationer af arvelig nefritis (med eller uden høretab) skyldes ekspressionsgraden af mutantgenet. Således betragtes i øjeblikket alle kliniske varianter som manifestationer af en sygdom, og udtrykket "arvelig nefritis" er synonymt med udtrykket "Alport syndrom".
Ifølge epidemiologiske undersøgelser forekommer arvelig nephritis med en hyppighed på 17 pr. 100.000 børn.
Årsager til Alport syndrom
Det genetiske grundlag for sygdommen er en mutation i gen a-5 i kollagenkæden af type IV. Denne type er universel for de basale membraner i nyren, cochlear, linsekapsel, nethinden og hornhinden i øjet, hvilket er bevist i undersøgelser, der anvender monoklonale antistoffer mod denne kollagenfraktion. For nylig angiver de muligheden for at anvende DNA-prober til prænatal diagnose af arvelig nefritis.
Betydningen af at teste alle familiemedlemmer ved hjælp af DNA-prober for at identificere bærere af mutantgenet er understreget, hvilket er af stor betydning for at udføre medicinsk genetisk rådgivning af familier med denne sygdom. Dog har op til 20% af familierne ikke familiemedlemmer med nyresygdom, hvilket tyder på en høj forekomst af spontane mutationer i det unormale gen. De fleste patienter med arvelig nefritis hos familier har personer med nyresygdom, høretab og synspatologi; relaterede ægteskaber mellem mennesker, der har en eller flere forfædre, da ægteskabet mellem beslægtede personer øger sandsynligheden for at opnå de samme gener fra begge forældre. Autosomal dominant og autosomal recessiv og dominant, der er forbundet med X-kromosomet af transmissionsvejen, etableres.
Børn er mere tilbøjelige til at skelne mellem tre varianter af arvelig nephritis: Alport syndrom, arvelig nefritis uden høretab og familiemæssig godartet hæmaturi.
Alports syndrom - arvelig nefritis med høreskader. Grundlaget er en kombineret defekt i strukturen af kollagen af den basale membran i glomeruli i nyrerne, øre og øje strukturer. Genet af det klassiske Alport syndrom er placeret på stedet 21-22 q af den lange arm af X-kromosomet. I de fleste tilfælde er det arvet af den dominerende type, der er knyttet til X-kromosomet. Hos mænd er Alport syndrom vanskeligere, fordi hos kvinder er mutantgenfunktionen kompenseret af en sund allel af det andet intakte kromosom.
Genetisk grundlag for udvikling af arvelig nefrit er mutationer i gener af alfa kæder af type IV kollagen. Seks a-kæder af type IV-kollagen er kendt: generne af a5- og a6-kæder (Co4A5 og Co4A5) er placeret på den lange arm af X-kromosomet i 21-22q-zonen; gener af a3- og a4-kæder (Co4A3 og Co4A4) - på 2-nd-kromosomet; gener af a1- og a2-kæder (Co4A1 og Co4A2) - på det 13. Chromosom.
I de fleste tilfælde (80-85%) er en X-koblet type sygdomarv forbundet med skade på Co4A5-genet på grund af deletion, punktmutationer eller splejsningsforstyrrelser. I øjeblikket findes mere end 200 mutationer af genet Kol4A5, der er ansvarlig for overtrædelsen af syntese af a5-kæder af kollagen type IV. I denne type arv manifesterer sig sygdommen hos børn af begge køn, men hos drenge er det vanskeligere.
Mutationer i loci af generne Co4A3 og Co4A4, der er ansvarlige for syntese af a3- og a4-kæder af type IV-kollagen, arves autosomalt. Ifølge forskning observeres autosomal dominerende arvstype i 16% af arvelige nefritis, autosomal recessiv - hos 6% af patienterne. Der er ca. 10 mutationer af generne Co4A3 og Co4A4.
Resultatet af mutationer er en overtrædelse af processerne ved samling af type IV kollagen, hvilket fører til en forstyrrelse i dens struktur. Kollagen type IV er en af hovedkomponenterne i den glomerulære basalmembran, det cochleære apparat og øjets linsen, hvis patologi afsløres i klinikken for arvelig nefritis.
Collagen type IV, del af den glomerulære basalmembran, består i det væsentlige af to kæder a1 (IV) og en a2-kæde (IV), og indeholder også a3, a4, a5 kæde. Oftest når X-bunden arv Sol4A5 mutation ledsaget af mangel a3, A4- og A6 A5 kæder af collagen type IV i strukturen, og antallet af O1 og a2 kæder i den glomerulære basalmembran stiger. Mekanismen for dette fænomen er uklart, det antages, at årsagen er posttransskriptionelle ændringer i mRNA.
Mangel a3, A4- og A5 kæder i strukturen type IV-collagen basismembranen i glomeruli resulterer i udtynding og skrøbelighed af de tidlige stadier af Alport-syndrom, der manifesterer sig klinisk fleste hæmaturi (undertiden hæmaturi eller proteinuri kun proteinuri), høretab og lenticonus. Yderligere progression af sygdommen fører til fortykkelse, og afbrydelse af basalmembranen permeabilitet i de sene stadier af sygdommen, med væksten i disse kollagentyper V og VI, manifesteret i stigningen af proteinuria og nedsatte nyrefunktion.
Naturen af mutationen underliggende arvelig nefritis bestemmer i vid udstrækning dens fænotypiske manifestation. Når X kromosom deletioner med samtidig mutation og Sol4A6 Sol4A5 gener er ansvarlige for syntesen af A5 og A6 kæder af collagen type IV, kombineret med Alports syndrom leiomyomatosis spiserøret og kønsorganer. Ifølge undersøgelser med Sol4A5 genmutationer forbundet med en deletion er mærket stor sværhedsgraden af den patologiske proces, en kombination med en renal læsion extrarenale manifestationer og tidlig udvikling af kronisk nyresvigt, sammenlignet stochechnoy mutation af dette gen.
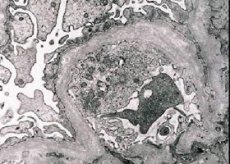
Morfologisk afslører elektronmikroskopi udtyndingen og delaminering af glomerulære basale membraner (især lamina densa) og tilstedeværelsen af elektronisk tynde granulater. Glomerulus læsionen kan være uensartet i den samme patient, fra minimal fokal læsion af mesangium til glomerulosklerose. Glomerulitis i Alport syndrom er altid immun-negativ, hvilket adskiller det fra glomerulonefritis. Karakteristisk er udviklingen af tubulær atrofi, lymfohistiocytinfiltrering, tilstedeværelsen af "skumceller" med inklusioner af lipider - lipofagi. Med sygdommens fremgang afsløres en fortykning og markant ødelæggelse af de basale glomeruli membraner.
Visse ændringer i immunsystemets tilstand afsløres. Hos patienter med arvelig nefrit er der konstateret et fald i niveauet af Ig A og en tendens til at øge koncentrationen af IgM i blodet, niveauet af IgG kan forøges i de tidlige stadier af sygdommens udvikling og fald i sent termer. Måske er en stigning i koncentrationen af IgM og G en slags kompenserende respons som reaktion på et underskud af IgA.
Den funktionelle aktivitet af T-lymfocyt-systemet reduceres; Det markerede selektiv reduktion af B-lymfocytter, der er ansvarlig for syntesen af Ig A, fordelt fagocytisk immunitet link, hovedsagelig på grund af nedbrydning af processer kemotaksi og intracellulær spaltning af neutrofiler
I undersøgelsen af nyrebiopsi hos patienter med Alports syndrom ved elektronmikroskopi, ultrastrukturelle ændringer observeret glomerulære basalmembran: udtynding, og opsplitning mønstre krænkelse glomerulære basalmembraner med ændringen i dets tykkelse og ujævne konturer. I de tidlige stadier af arvelig nefritis bestemmer defekten fortynding og skrøbelighed af de glomerulære basale membraner.
Tyndning af glomerulære membraner er et mere gunstigt tegn og er mere almindeligt hos piger. En mere konstant elektronmikroskopisk funktion i arvelig nefritis er spaltningen af den basale membran, og sværhedsgraden af dens ødelæggelse korrelerer med procesens sværhedsgrad.

Symptomer på Alports syndrom hos børn
De første symptomer på Alport syndrom i form af isoleret urinssyndrom registreres oftere hos børn i de første tre år af livet. I de fleste tilfælde registreres sygdommen ved et uheld. Urinsyndromet afsløres under forebyggende undersøgelse af barnet, før de kommer ind i børns institution eller under ARVI. Ved forekomst af patologi i urinen under ARVI. I arvelig nefritis er der i modsætning til den erhvervede glomerulonefritis ingen latent periode.
I sygdommens indledende fase lider barnets velvære lette, det karakteristiske træk er persistensen og vedholdenheden af urinsyndromet. Et af hovedtegnene er hæmaturi af forskellig grad, observeret i 100% af tilfældene. Forøgelsen af graden af hæmaturi ses under eller efter luftvejsinfektioner, fysisk anstrengelse eller efter forebyggende vaccinationer. Proteinuri i de fleste tilfælde ikke overstiger 1 g / dag, ved sygdomsudbruddet kan det være ustabilt, da processen skrider frem, proteinurien øges. Periodisk set kan urinsedimentet have leukocyturi med en overvejelse af lymfocytter, der er forbundet med udviklingen af interstitiale forandringer.
Senere er der en krænkelse af nyres delvise funktioner, forværring af patientens generelle tilstand: forgiftning, muskelsvaghed, arteriel hypotension, ofte nedsat hørelse (især hos drenge), nogle gange nedsat syn. Intoxikation manifesterer sig som plager, træthed, hovedpine. I den første fase af sygdommen opdages høretab i de fleste tilfælde kun ved audiografi. Høretab i Alport syndrom kan forekomme i forskellige perioder med barndom, men oftest er høretab diagnosticeret i en alder af 6-10 år. Høretabet hos børn begynder ved høje frekvenser og når en betydelig grad i luft- og benledning, der går fra lydledende til lydmodtagende døvhed. Høretab kan være et af de første symptomer på sygdommen og kan foregribe urinsyndrom.
I 20% af tilfældene har patienter med Alport syndrom ændringer i øjnene. De mest almindelige anomalier fra linsen: spherofokiya, lentikonus anterior, posterior eller blandet, en række katarakter. I familier med Alport syndrom er der en betydelig forekomst af nærsynethed. En række forskere konstant i disse familier noterer sig bilaterale perimakulære ændringer i form af lyse hvide eller gullige granuleringer i området med den gule krop. De betragter dette symptom som et konstant symptom, som har høj diagnostisk værdi i Alport syndrom. C. S. Chugh et al. (1993) til oftalmologisk undersøgelse viste Alport syndrom patienter nedsat synsskarphed i 66,7% af tilfældene, den forreste lenticonus - 37,8%, pletterne på nethinden - i 22,2%, grå stær - 20%, keratoconus - 6 , 7%.
Hos nogle børn med arvelig nefritis, især i dannelsen af nyreinsufficiens, er der konstateret en signifikant forsinkelse i fysisk udvikling. Som udviklingen af nyreinsufficiens udvikler hypertension. Hos børn opdages det oftere hos unge og i ældre aldersgrupper.
Karakteristisk er tilstedeværelsen hos patienter med arvelig nefritis af forskellige (mere end 5-7) stigmas af bindevævsdysembryogenese. Blandt de bindevæv af stigmatisering i patienter med den hyppigste øje hypertelorism, høj gane, malocclusion, unormal form af ørerne, krumning den lille finger på hans hænder, "sandalevidnaya gap" på fødderne. For arvelig nefritis er kendetegnet ved ensartethed dizembriogeneza stigma i familien, såvel som den høje frekvens af deres fordeling i probander slægtninge, hvorigennem sygdommen overføres.
I de tidlige stadier af sygdommen afslørede en isoleret reduktion af delvis nyrefunktion: transport af aminosyrer, elektrolytter, koncentration funktioner Syredannelse, yderligere ændringer er funktionelle tilstand af både den proximale og distale nephron og har karakter af kombinerede partielle lidelser. Reduktion af glomerulær filtrering forekommer senere, oftere i ungdomsperioden. Efterhånden som arvelige nefritis udvikler sig, udvikler anæmi.
Således for arvelig nefritis kendetegnet stadieinddeling af sygdom: første latent stadium eller skjulte kliniske symptomer manifesteret ved minimale ændringer blæresyndrom sker derefter gradvis dekompensation proces med en reduktion i nyrefunktion med åbenlyse kliniske symptomer (forgiftning, asteni, udviklingsmæssige forsinkelser, anemizatsiya). Kliniske symptomer forekommer normalt uanset stratifikation af den inflammatoriske reaktion.
Arvelig nefritis kan manifestere sig i forskellige alder, hvilket afhænger af genets handling, som indtil en bestemt tid er i en undertrykt tilstand.
Klassifikation
Der er tre varianter af arvelig nefritis
- Jeg variant - er klinisk manifesteret af nefritis med hæmaturi, høretab og øjenskade. Nephritisforløbet er progressivt med udviklingen af CRF. Arvstypen er dominerende, knyttet til X-kromosomet. Morfologisk er der en forstyrrelse af strukturen af basalmembranen, dens udtynding og spaltning.
- II-variant-er klinisk manifesteret af nefritis med hæmaturi uden høretab. Nephritisforløbet er progressivt med udviklingen af kronisk nyresvigt. Arvstypen er dominerende, knyttet til X-kromosomet. Morfologisk afsløres udtynding af den basale membran af glomerulære kapillærer (især laminadensa).
- III mulighed - godartet familie hæmaturi. Kurset er gunstigt, kronisk nyresvigt udvikler sig ikke. Arvstypen er autosomal dominant eller autosomal recessiv. I den autosomale recessive type arv har kvinder et mere alvorligt forløb af sygdommen.
Diagnose af Alport syndrom
Følgende kriterier foreslås:
- tilstedeværelsen i hver familie af mindst to patienter med nefropati
- hæmaturi som det førende symptom på nefropati i proband;
- mindst et medlem af familien har et høretab
- udvikling af kronisk nyresvigt i en relativ og mere.
I diagnosticering af en række arvelige og medfødte sygdomme en vigtig plads tilhører en integreret tilgang til inspektion og frem for alt at være opmærksom på de data, der er opnået i forbindelse med udarbejdelsen af barnets stamtavle. Diagnose syndrom Alport anses for gyldige i tilfælde, hvor patienten 3 ud af 4 typiske træk: tilstedeværelsen i familien hæmaturi og kronisk nyresvigt, tilstedeværelsen af patientens sensorineuralt høretab, patologier af påvisning i elektronmikroskopisk karakterisering biopsi tegn spaltning glomerulær basalmembran med en ændring af dens tykkelse og ujævne konturer.
Undersøgelsen af patienten bør omfatte kliniske genetiske undersøgelsesmetoder; rettet undersøgelse af sygdomens anamnese Generel undersøgelse af patienten under hensyntagen til diagnostiske kriterier. I kompensationsfasen kan man kun fange patologi ved at fokusere på sådanne syndromer som tilstedeværelsen af arvelig komplikation, hypotension, multipel stigmatisk dysembryogenese, ændringer i urinsyndromet. I dekompensationstrinnet kan der forekomme østrænale symptomer, såsom alvorlig forgiftning, asteni, forsinkelse i fysisk udvikling, anæmi, manifesteret og forstærket med et gradvist fald i nyrefunktionen. Hos de fleste patienter med nedsat nyrefunktion observeres et fald i funktionen af acido- og aminogenese; hos 50% af patienterne opdages et signifikant fald i sekretoriske funktion af nyrerne; begrænsning af svingninger i urinens optiske tæthed en krænkelse af filtreringens rytme og derefter et fald i glomerulær filtrering. Stadiet for kronisk nyresvigt diagnosticeres hos patienter med 3-6 måneder og mere forhøjet urinstofniveau i blodserum (mere end 0,35 g / l), et fald i glomerulær filtrering til 25% af normen.
Differentialdiagnose af arvelig nefritis skal udføres primært med den erhvervede form hematuric glomerulonephritis. Har fået stadig akut glomerulonephritis begyndende 2-3 uger periode efter en tidligere infektion, extrarenale funktioner, herunder hypertension med de første dage (ved arvelig nephritis omvendt hypotension), nedsat glomerulær filtrationshastighed ved debut, ingen brud på de partielle rørformede funktioner, hvorimod som med arvelige er de til stede. Erhvervet glomerulonephritis opstår med mere alvorlige hæmaturi og proteinuri, med øget ESR. Diagnostisk værdi er typiske ændringer i glomerulær basalmembran, karakteristisk arvelig nefritis.
Differentialdiagnose af dysmetaboliske nefropati udført med kronisk nyresvigt i familien identificeret klinisk monotypemaskiner nyresygdom, og kan variere fra nefropati pyelonephritis til urolithiasis. Børn har ofte klager over mavesmerter og jævnligt med vandladning, i urinsedimentet - oxalat.
Hvis du har mistanke om en arvelig nefritis, skal patienten sendes for at afklare diagnosen i en specialiseret nefrologisk afdeling.
Hvad skal man undersøge?
Hvordan man undersøger?
Hvilke tests er nødvendige?
Hvem skal kontakte?
Behandling af Alport syndrom
I regimet sørger for en begrænsning af stor fysisk anstrengelse, ophold i frisk luft. Kosten er høj kvalitet, med tilstrækkeligt indhold af højkvalitets proteiner, fedtstoffer og kulhydrater under hensyntagen til nyrernes funktion. Af stor betydning er identifikation og rehabilitering af kroniske infektionsfokus. Fra lægemidler anvendes carnitinchlorid, ATP, cocarboxylase, pyridoxin (op til 50 mg / dag). Kurserne afholdes 2-3 gange om året. Når hæmaturi er ordineret phytotherapy - nælde, nældebrune, blackberry aske, yarrow.
I fremmed og indenlandsk litteratur er der rapporter om behandling med prednisolon og brug af cytostatika. Effekten er dog svært at bedømme.
Ved kronisk nyresvigt anvendes hæmodialyse og nyretransplantation.
Der findes ingen metoder til specifik (effektiv patogenetisk) behandling af arvelig nefritis. Alle medicinske foranstaltninger tager sigte på at forebygge og nedsætte nedsættelsen af nyrefunktioner.
Kosten skal være afbalanceret og højkalorier under hensyntagen til nyrernes funktionelle tilstand. I mangel af krænkelser af den funktionelle tilstand i barnets ernæring bør der være et tilstrækkeligt indhold af proteiner, fedtstoffer og kulhydrater. I tilstedeværelsen af tegn på nyresvigt bør mængden af protein, kulhydrater af calcium og fosfor begrænses, hvilket forsinker udviklingen af kronisk nyresvigt.
Fysisk stress bør begrænses, børn anbefales at afstå fra at lave sport.
Undgå kontakt med infektiøse patienter, reducer risikoen for udvikling af akutte åndedrætsinfektioner. Det er nødvendigt at sanitere foci for kronisk infektion. Forebyggende vaccinationer til børn med arvelig nefritis udføres ikke, vaccination er kun mulig i henhold til epidemiologiske indikationer.
Hormonal og immunosuppressiv terapi i arvelig nephritis er ineffektiv. Der er tegn på en vis positiv effekt (et fald i proteinuriens niveau og en afmatning i sygdommens fremgang) med langvarig brug af cyclosporin A- og ACE-hæmmere i mange år.
Ved behandling af patienter, der bruger stoffer, der forbedrer stofskiftet:
- pyridoxin - 2-3 mg / kg / dag i 3 opdelte doser i 4 uger;
- kokarboksilaza - 50 mg intramuskulært hver anden dag, kun 10-15 injektioner;
- ATP - 1 ml intramuskulært hver anden dag, 10-15 injektioner;
- Vitamin A - 1000 U / år / dag i 1 modtagelse i 2 uger;
- E-vitamin - 1 mg / kg / dag i 1 modtagelse i 2 uger.
En sådan terapi forbedrer patientens generelle tilstand, reducerer tubulær dysfunktion og administreres 3 gange om året.
Da en immunmodulator kan anvendes levamisol - 2 mg / kg / dag 2-3 gange om ugen med intermissions mellem doser på 3-4 dage.
Til forskerne har hyperbarisk oxygenation en positiv effekt på sværhedsgraden af hæmaturi og nedsat nyrefunktion.
Den mest effektive metode til behandling af arvelig nefritis er rettidig nyretransplantation. Der er ingen tilbagevenden af sygdommen i transplantationen, i en lille procentdel af tilfælde (ca. 5%) er udviklingen af nefritis i den transplanterede nyre forbundet med antigener til den glomerulære basalmembran mulig.
Et lovende område er prænatal diagnose og genteknisk terapi. Eksperimenter på dyr viser en høj effektivitet ved overførsel af normale gener ansvarlig for syntesen af a-kæder af type IV-kollagen til nyrevævet, hvorefter syntese af normale kollagenstrukturer er noteret.
Outlook
Prognosen for arvelig nefritis er altid alvorlig.
Prognostisk ugunstige kriterier for strømmen af arvelig nefrit er:
- mandlige køn;
- tidlig udvikling af kronisk nyresvigt hos familiemedlemmer
- proteinuri (mere end 1 g / dag);
- fortykkelse af glomerulære basale membraner ifølge mikroskopi;
- neuritis af den auditive nerve;
- deletion i genet Co4A5.
Prognosen for godartet familiens hæmaturi er mere gunstig.
Использованная литература