Medicinsk ekspert af artiklen

Nye publikationer
Usher syndrom
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Usher syndrom er en arvelig sygdom, der manifesterer sig som fuldstændig døvhed fra fødslen, samt progressiv blindhed med alderen. Synstab er forbundet med retinitis pigmentosa, en proces med pigmentdegeneration af nethinden. Mange mennesker med Usher syndrom har også alvorlige balanceproblemer.
Epidemiologi
Takket være forskningen var det muligt at fastslå, at Usher syndrom rammer omkring 8% af de undersøgte døvstumme børn (testene blev udført på særlige institutioner for døvstumme). Pigmentær retinitis blev observeret hos 6-10% af patienter, der lider af medfødt døvhed, hvilket igen observeres hos omkring 30% af personer med pigmenteret nethindesygdom.
Det menes, at denne sygdom manifesterer sig hos cirka 3-10 personer ud af 100.000 på verdensplan. Den kan observeres ligeligt hos både kvinder og mænd. Omkring 5-6% af verdens befolkning lider af dette syndrom. Omkring 10% af alle tilfælde af dyb døvhed i børneårene skyldes Usher syndrom I, såvel som II typer.
I USA er type 1 og 2 de mest almindelige typer. Tilsammen tegner de sig for cirka 90 til 95 procent af alle tilfælde af Usher syndrom hos børn.
Årsager Usher syndrom
Usher syndrom type I, II og III har en autosomal recessiv årsag, mens type IV betragtes som en X-kromosomforstyrrelse. Årsagerne til blindhed og døvhed, der opstår ved dette syndrom, er endnu ikke tilstrækkeligt undersøgt. Det antages, at personer med denne sygdom er overfølsomme over for komponenter, der kan beskadige DNA-strukturen. Derudover kan denne sygdom være forbundet med immunsystemforstyrrelser, men i dette tilfælde er der ikke noget præcist billede af denne proces.
I 1989 blev kromosomafvigelser først identificeret hos patienter med type II-sygdom, hvilket i fremtiden kan føre til en metode til at isolere de gener, der forårsager syndromet. Det kan også være muligt at identificere disse gener hos bærere og udvikle særlige prænatale genetiske tests.
 [ 8 ]
[ 8 ]
Risikofaktorer
Syndromet nedarves, når begge forældre er berørt, dvs. det nedarves af en recessiv type. Et barn kan også arve sygdommen, hvis forældrene er bærere af genet. Hvis begge kommende forældre har dette gen, er sandsynligheden for at få et barn med syndromet 1 til 4. En person, der kun har ét gen for syndromet, betragtes som bærer, men ikke har symptomer på lidelsen. I dag er det endnu ikke muligt at afgøre, om en person har genet for denne sygdom.
Hvis et barn fødes af forældre, hvoraf den ene ikke har et sådant gen, er sandsynligheden for, at han arver syndromet, meget lav, men han vil helt sikkert være bærer.
Symptomer Usher syndrom
Symptomer på Usher syndrom omfatter høretab og unormal ophobning af pigmenterede celler i øjets strukturer. Patienten udvikler derefter degeneration af nethinden, hvilket forårsager synsforringelse og i sidste ende synstab i de mest alvorlige tilfælde.
Sensorineuralt høretab kan være mildt eller fuldstændigt og udvikler sig normalt ikke fra fødslen. Imidlertid kan retinapigmentsygdom begynde at udvikle sig i barndommen eller senere. Testresultater har vist, at den centrale synsstyrke kan opretholdes i mange år, selv når det perifere syn forringes (en tilstand kaldet "tunnelsyn").
Dette er sygdommens primære manifestationer, som undertiden kan suppleres af andre lidelser, såsom psykose og andre psykiske lidelser, problemer med det indre øre og/eller grå stær.
Forms
Under forskningen blev der identificeret 3 typer af denne sygdom, samt en fjerde form, som er ret sjælden.
Type I af sygdommen er karakteriseret ved medfødt fuldstændig døvhed samt balanceforstyrrelser. Ofte begynder sådanne børn først at gå i en alder af 1,5 år. Forværring af synet begynder normalt i 10-årsalderen, og den endelige udvikling af tilstanden natblindhed begynder i 20-årsalderen. Børn med denne type sygdom kan udvikle en progressiv forværring af det perifere syn.
Ved type II-sygdom observeres moderat eller medfødt døvhed. I dette tilfælde ophører forværringen af delvis døvhed ofte med at forekomme. Pigmentær retinitis begynder at udvikle sig omkring slutningen af ungdomsårene eller efter 20 år. Udviklingen af natteblindhed begynder normalt i alderen 29-31 år. Synsnedsættelse ved type II-patologi forløber generelt lidt langsommere end ved type I.
Type III af sygdommen er karakteriseret ved progressivt høretab, der normalt begynder i puberteten, samt den gradvise udvikling i samme periode (lidt senere end høretabet) af retinitis pigmentosa, hvilket kan blive en faktor i udviklingen af progressiv blindhed.
Manifestationer af type IV-patologi forekommer hovedsageligt hos mænd. I dette tilfælde observeres også progressive lidelser og tab af hørelse og syn. Denne form er meget sjælden og har normalt en X-kromosomal natur.
Diagnosticering Usher syndrom
Diagnosen Usher syndrom stilles ud fra patientens observerede kombination af pludselig døvhed og progressivt synstab.
Test
En særlig genetisk test kan bestilles for at opdage mutationen.
Der er fundet elleve genetiske loci, der kan forårsage udviklingen af Usher syndrom, og ni gener er blevet identificeret, der med sikkerhed er årsagen til lidelsen:
- Type 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Type 2: ush2a, VLGR1, WHRN.
- Usher syndrom type 3: USH3A.
NIDCD-forskere har sammen med kolleger fra universiteter i New York og Israel identificeret en mutation kaldet R245X i Pcdh15-genet, der tegner sig for en stor procentdel af type 1 Usher syndrom i den jødiske befolkning.
For at finde ud af mere om laboratorier, der udfører kliniske forsøg, kan du besøge https://www.genetests.org og søge i laboratoriekataloget efter "Usher syndrom".
For at lære om eksisterende kliniske forsøg, der inkluderer genetisk testning for Usher syndrom, kan du besøge https://www.clinicaltrials.gov og søge efter "Usher syndrom" eller "Usher syndrome genetic testing".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentel diagnostik
Der er flere metoder til instrumentel diagnostik:
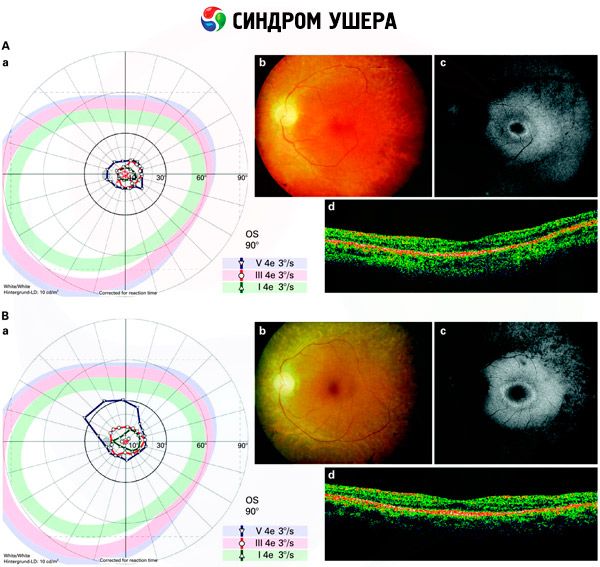
- Undersøgelse af fundus for at detektere tilstedeværelsen af pigmentpletter på nethinden, samt indsnævring af nethindekarrene;
- Elektroretinogram, som gør det muligt at detektere indledende degenerative afvigelser i øjets nethinde. Det viser udslettelsen af elektroradiografiske signalveje;
- Et elektronystagmogram (ENG) måler ufrivillige øjenbevægelser, der kan indikere tilstedeværelsen af en ubalance.
- Audiometri, som bruges til at bestemme tilstedeværelsen af døvhed og dens sværhedsgrad.
Differential diagnose
Usher syndrom skal differentieres fra nogle lignende lidelser.
Hallgrens syndrom, som er karakteriseret ved medfødt høretab og progressivt synstab (grå stær og nystagmus udvikles også). Yderligere symptomer omfatter ataksi, psykomotoriske forstyrrelser, psykose og mental retardering.
Alstrom syndrom, som er en arvelig sygdom, hvor nethinden degenererer, hvilket resulterer i tab af centralt syn. Dette syndrom er forbundet med børnefedme. Samtidig begynder diabetes mellitus og høretab at udvikle sig efter 10 år.
Røde hunde hos en gravid kvinde i første trimester kan forårsage forskellige abnormiteter i barnets udvikling. Blandt konsekvenserne af en sådan abnormitet er høretab, såvel som (eller) problemer med synet, og derudover forskellige udviklingsdefekter.
Hvem skal kontakte?
Behandling Usher syndrom
Der findes i øjeblikket ingen kur mod Usher syndrom. Derfor består behandlingen i dette tilfælde primært af at bremse processen med synstab samt at kompensere for høretab. Mulige behandlingsmetoder omfatter:
- Indtagelse af A-vitamin (nogle øjenlæger mener, at høje doser af A-vitaminpalmitat kan bremse, men ikke stoppe, udviklingen af retinitis pigmentosa);
- Implantation af specielle elektroniske apparater i patientens ører (høreapparater, cochlearimplantater).
Øjenlæger anbefaler, at de fleste voksne med almindelige former for retinitis pigmentosa tager 15.000 IE (internationale enheder) vitamin A-palmitat dagligt under opsyn. Da personer med type 1 Usher syndrom ikke var inkluderet i undersøgelsen, anbefales høje doser af vitamin A ikke til denne patientgruppe. Personer, der overvejer at tage vitamin A, bør diskutere denne behandlingsmulighed med deres læge. Andre anbefalinger til denne behandlingsmulighed inkluderer:
- Ændring af din kost til at inkludere fødevarer med et højt indhold af A-vitamin.
- Kvinder, der planlægger at blive gravide, bør stoppe med at tage høje doser af A-vitamin tre måneder før, de planlægger at blive gravide, på grund af en øget risiko for fosterskader.
- Gravide kvinder bør stoppe med at tage høje doser af A-vitamin på grund af en øget risiko for fosterskader.
Det er også vigtigt at tilpasse et sådant barn til det sociale liv. Dette kræver hjælp fra specialpædagogiske lærere og psykologer. I tilfælde af at patienten er begyndt at opleve progressivt synstab, bør han lære at bruge tegnsprog.
Vejrudsigt
Usher syndrom har en ugunstig prognose. Synsfeltet og dets skarphed begynder at forringes i perioden 20-30 år hos de fleste patienter med denne sygdom af enhver type. I nogle tilfælde forekommer fuldstændigt bilateralt synstab. Høretab, som altid ledsages af sløvhed, udvikler sig meget hurtigt til fuldstændigt bilateralt høretab.