Medicinsk ekspert af artiklen

Nye publikationer
Charcot-Marie-Tooth sygdom.
Sidst revideret: 12.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
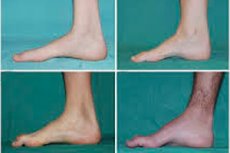
Peroneal muskelatrofi, Charcot-Marie-Tooth syndrom eller sygdom er en gruppe af kroniske arvelige sygdomme med skader på de perifere nerver.
Ifølge ICD-10, i afsnittet om sygdomme i nervesystemet, er koden for denne sygdom G60.0 (arvelig motorisk og sensorisk neuropati). Den er også inkluderet på listen over sjældne sygdomme.
Epidemiologi
Ifølge klinisk statistik er forekomsten af alle typer Charcot-Marie-Tooth sygdom pr. 100 tusinde af befolkningen 19 tilfælde (ifølge andre kilder, et tilfælde pr. 2,5-10 tusinde af befolkningen).
CMT type 1 tegner sig for omkring to tredjedele af tilfældene (et tilfælde pr. 5.000 til 7.000 indbyggere), og næsten 70% af dem er forbundet med duplikering af PMP22-genet. Mere end 1,2 millioner mennesker verden over lider af denne type sygdom.
Forekomsten af CMT type 4 er anslået til 1-5 tilfælde pr. 10.000 børn. [ 1 ]
Årsager Charcot-Marie-Tooth sygdom
Ifølge klassificeringen af polyneuropatiske syndromer refererer peroneal (fibulær) muskelatrofi, Charcot-Marie-Tooth neural amyotrofi eller Charcot-Marie-Tooth sygdom (forkortet CMT) til genetisk bestemte motorisk-sensoriske polyneuropatier. [ 2 ]
Det vil sige, at årsagerne til dens forekomst er genetiske mutationer. Og afhængigt af arten af genetiske afvigelser skelnes der mellem hovedtyperne eller -typerne af dette syndrom: demyeliniserende og aksonale. Den første gruppe omfatter Charcot-Marie-Tooth sygdom type 1 (CMT1), som opstår på grund af duplikering af PMP22-genet på kromosom 17, som koder for transmembran perifer myelinprotein 22. Som følge heraf forekommer segmental demyelinisering af axonskeden (nervecelleprocesser) og et fald i hastigheden af nervesignalledning. Derudover kan mutationer forekomme i nogle andre gener.
Den aksonale form er Charcot-Marie-Tooth sygdom type 2 (CMT2), som påvirker selve axonerne og er forbundet med patologiske ændringer i MFN2-genet på locus 1p36.22, der koder for membranproteinet mitofusin-2, som er nødvendigt for mitokondriefusion og dannelsen af funktionelle mitokondrielle netværk inde i perifere nerveceller. Der er mere end et dusin undertyper af CMT2 (med mutationer i specifikke gener).
Det skal bemærkes, at der i øjeblikket er identificeret mere end hundrede gener, hvis skade, der overføres ved arv, forårsager forskellige undertyper af Charcot-Marie-Tooth sygdom. For eksempel fører mutationer i RAB7-genet til CMT type 2B; ændring af SH3TC2-genet (som koder for et af membranproteinerne i Schwann-celler) forårsager CMT type 4C, som manifesterer sig i barndommen og er karakteriseret ved demyelinisering af motoriske og sensoriske neuroner (der er et dusin og et halvt former for type 4 af denne sygdom).
En sjælden type 3 CMT (kaldet Dejerine-Sottas syndrom) begynder at udvikle sig i den tidlige barndom og er forårsaget af mutationer i PMP22, MPZ, EGR2 og andre gener.
Når CMT type 5 opstår i alderen 5-12 år, observeres ikke kun motorisk neuropati (i form af spastisk paraparese i underekstremiteterne), men også skader på syns- og hørenerverne.
Muskelsvaghed og optisk atrofi (med synstab) samt balanceproblemer er typiske for CMT type 6. Og ved type 7 Charcot-Marie-Tooth sygdom er der ikke kun motorisk-sensorisk neuropati, men også en nethindesygdom i form af retinitis pigmentosa.
X-bundet CMT eller Charcot-Marie-Tooth sygdom med tetraparese af lemmerne (bevægelsessvaghed i både arme og ben), som er mere almindelig blandt mænd, er en demyeliniserende type og menes at skyldes en mutation i GJB1-genet på den lange arm af X-kromosomet, som koder for connexin 32, et transmembranprotein i Schwann-celler og oligodendrocytter, der regulerer transmissionen af nervesignaler. [ 3 ]
Risikofaktorer
Den primære risikofaktor for CMT er en familiehistorie med sygdommen, det vil sige hos nære slægtninge.
Ifølge genetikere er risikoen for at få et barn, der udvikler sygdommen, 25 %, hvis begge forældre er bærere af det autosomalt recessive gen for Charcot-Marie-Tooths sygdom, og risikoen for, at barnet er bærer af dette gen (men ikke har nogen symptomer), anslås til 50 %.
I tilfælde af X-bundet arv (når det muterede gen sidder på kvindens X-kromosom) er der en 50% risiko for, at moderen giver genet videre til sin søn, som vil udvikle CMT. Sygdommen opstår muligvis ikke, når et pigebarn fødes, men datterens sønner (børnebørn) kan arve det defekte gen, og sygdommen vil udvikle sig.
Patogenese
I enhver type Charcot-Marie-Tooth sygdom er dens patogenese forårsaget af en arvelig anomali i de perifere nerver: motoriske (bevægelse) og sensoriske (følsomme).
Hvis CMT-typen er demyeliniserende, fører ødelæggelsen eller defekten af myelinskeden, der beskytter axonerne i de perifere nerver, til en afmatning i transmissionen af nerveimpulser i det perifere nervesystem - mellem hjernen, musklerne og sanseorganerne.
I den aksonale type af sygdommen påvirkes selve axonerne, hvilket negativt påvirker styrken af nervesignaler, hvilket er utilstrækkeligt til fuld stimulering af muskler og sanseorganer.
Læs også:
Hvordan overføres Charcot-Marie-Tooth syndrom? De defekte gener kan nedarves autosomalt dominant eller autosomalt recessivt.
Den mest almindelige type, autosomal dominant arv, forekommer, når der er én kopi af det muterede gen (båret af en af forældrene). Og sandsynligheden for at give CMT videre til hvert født barn er anslået til 50%. [ 4 ]
Ved autosomal recessiv arv kræves der to kopier af det defekte gen (en fra hver forælder, der ikke viser tegn på sygdommen), for at sygdommen kan udvikle sig.
I 40-50% af tilfældene forekommer autosomal dominant arvelig demyelinisering, dvs. CMT type 1; i 12-26% af tilfældene forekommer axonal CMT, dvs. type 2. Og i 10-15% af tilfældene observeres X-bundet arv. [ 5 ]
Symptomer Charcot-Marie-Tooth sygdom
Typisk begynder de første tegn på denne sygdom at vise sig i barndommen og ungdomsårene og udvikler sig gradvist gennem hele livet, selvom syndromet kan vise sig senere. Kombinationen af symptomer er variabel, og sygdommens progressionshastighed samt dens sværhedsgrad er umulig at forudsige.
Typiske symptomer på den indledende fase inkluderer øget generel træthed; nedsat tonus (svaghed) i musklerne i fødder, ankler og skinneben; mangel på reflekser. Dette gør fodbevægelse vanskelig og fører til dysbasi (gangforstyrrelser) i form af en højere benløftning, ofte med hyppige snublen og fald. Tegn på Charcot-Marie-Tooth sygdom hos et lille barn kan omfatte udtalt klodsethed og aldersukarakteristiske gangvanskeligheder forbundet med bilateral dropfod. Foddeformiteter er også karakteristiske: høj bue (hulfod) eller alvorlig platfod, krumme (hammerformede) tæer.
I tilfælde af at barnet går på tæerne på baggrund af muskelhypotoni, kan en neurolog have mistanke om, at barnet har type 4 CMT, hvor børn muligvis ikke kan gå i ungdomsårene.
Efterhånden som sygdommen skrider frem, spreder muskelatrofi og svaghed sig til de øvre ekstremiteter, hvilket gør det vanskeligt at udføre finmotorik og normale håndopgaver. Nedsat taktil fornemmelse og evnen til at føle varme og kulde, samt følelsesløshed i fødder og hænder, indikerer skader på axonerne i de følende nerver.
Ved Charcot-Marie-Tooth sygdom type 3 og 6, der manifesterer sig i barndommen, observeres sensorisk ataksi (nedsat koordination af bevægelser og balance), muskeltrækninger og tremor, skade på ansigtsnerven, atrofi af synsnerven med nystagmus og høretab.
I senere stadier kan der være ukontrollerbar rysten (tremor) og hyppige muskelkramper; bevægelsesproblemer kan føre til udvikling af smerter: muskel-, led-, neuropatisk.
Komplikationer og konsekvenser
Charcot-Marie-Tooths sygdom kan have komplikationer og konsekvenser såsom:
- hyppigere forstuvninger og brud;
- kontrakturer forbundet med forkortelse af periartikulære muskler og sener;
- skoliose (krumning af rygsøjlen);
- vejrtrækningsproblemer – når nervefibrene, der innerverer musklerne i mellemgulvet, er beskadiget:
- tab af evnen til at bevæge sig selvstændigt.
Diagnosticering Charcot-Marie-Tooth sygdom
Diagnosen omfatter klinisk undersøgelse, anamnese (inklusive familiehistorie), neurologisk og systemisk undersøgelse.
Der udføres tests for at kontrollere bevægelsesomfang, følsomhed og senereflekser. Nerveledningsevnen kan vurderes ved hjælp af instrumentel diagnostik – elektromyografi eller elektroneuromyografi. Ultralyd eller MR kan også være nødvendig. [ 6 ]
Genetiske eller DNA-tests til at detektere de mest almindelige genetiske mutationer, der forårsager CMT i en blodprøve, er begrænsede, fordi DNA-tests i øjeblikket ikke er tilgængelige for alle typer af sygdommen. For mere information, se Genetiske tests.
I nogle tilfælde udføres en biopsi af den perifere nerve (normalt den surale nerve).
Differential diagnose
Differentialdiagnose omfatter andre perifere neuropatier, Duchennes muskeldystrofi, myelopatiske og myastheniske syndromer, diabetisk neuropati, myelopatier ved multipel og amyotrofisk lateral sklerose, Guillain-Barré syndrom, traume på peronealnerven og dens atrofi (herunder ved klemning mellem lændeskiverne i rygsøjlen), skade på lillehjernen eller thalamus, samt bivirkninger af kemoterapi (under behandling med cytostatika såsom Vincristin eller Paclitaxel). [ 7 ]
Hvem skal kontakte?
Behandling Charcot-Marie-Tooth sygdom
I dag omfatter behandlingen af denne arvelige sygdom træningsterapi (der har til formål at styrke og strække musklerne); ergoterapi (som hjælper patienter med muskelsvaghed i hænderne); og brug af ortopædiske hjælpemidler til at lette gang. Om nødvendigt ordineres smertestillende eller antikonvulsive midler. [ 8 ]
I tilfælde af svære platfodsproblemer kan osteotomi udføres, og i tilfælde af hældeformation er kirurgisk korrektion indiceret – artrodese. [ 9 ]
Der forskes i både sygdommens genetiske komponent og dens behandlingsmetoder. Brugen af stamceller, visse hormoner, lecithin eller ascorbinsyre har endnu ikke givet positive resultater.
Men takket være nyere forskning kan der faktisk dukke noget nyt op i behandlingen af Charcot-Marie-Tooths sygdom i den nærmeste fremtid. Således har det franske firma Pharnext siden 2014 udviklet, og siden midten af 2019 har kliniske forsøg været i gang med lægemidlet PXT3003 til behandling af CMT type 1 hos voksne, hvilket undertrykker øget ekspression af PMP22-genet, forbedrer myelinisering af perifere nerver og lindrer neuromuskulære symptomer.
Specialister fra medicinalfirmaet Sarepta Therapeutics (USA) arbejder på at skabe en genterapi mod Charcot-Marie-Tooth sygdom type 1. Denne terapi vil bruge en harmløs adenoassocieret virus (AAV) af Dependovirus-slægten med et lineært enkeltstrenget DNA-genom, som vil overføre NTF3-genet til kroppen, der koder for neurotrophin-3 (NT-3)-proteinet, der er nødvendigt for Schwann-nervecellernes funktion.
Helixmith vil begynde kliniske forsøg med den sydkoreanskudviklede genterapi Engensis (VM202) inden udgangen af 2020 til behandling af muskelsymptomer ved CMT type 1. [ 10 ]
Forebyggelse
Forebyggelse af CMT kan være genetisk rådgivning af kommende forældre, især hvis en person i parret har en familiehistorie med sygdommen. Der er dog identificeret tilfælde af de novo punktgenmutationer, det vil sige i fravær af sygdommen i familiens historie.
Under graviditeten kan sandsynligheden for Charcot-Marie-Tooth sygdom hos det fremtidige barn kontrolleres ved hjælp af en chorionvillusbiopsi (fra 10. til 13. graviditetsuge) samt en analyse af fostervand (ved 15-18. uge).
Vejrudsigt
Generelt afhænger prognosen for forskellige typer Charcot-Marie-Tooth sygdom af den kliniske sværhedsgrad, men i alle tilfælde udvikler sygdommen sig langsomt. Mange patienter har handicap, selvom dette ikke reducerer den forventede levetid.