Nye publikationer
Trigonocephali
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
En medfødt anomali i form af en kraniedeformation, hvor spædbørns hoved har en uregelmæssig form, og kraniet fremstår trekantet, defineres som trigonocephali (fra græsk trigonon – trekant og kephale – hoved). [ 1 ]
Epidemiologi
Prævalensen af kraniosynostose anslås til cirka fem tilfælde pr. 10.000 levendefødte (eller ét tilfælde pr. 2.000-2.500 personer i den generelle befolkning). [ 2 ]
I 85% af tilfældene er kraniosynostose sporadisk, de resterende tilfælde forekommer som en del af et syndrom. [ 3 ]
Ifølge statistikker er for tidlig fusion af den mediane frontale sutur den næstmest almindelige form for kraniosynostose, og trigonocephali tegner sig for ét tilfælde ud af 5-15 tusind nyfødte; antallet af drengebørn med denne anomali er næsten tre gange højere end antallet af pigebørn. [ 4 ]
I cirka 5% af tilfældene er denne medfødte anomali til stede i familiens historie. [ 5 ]
Årsager trigonocephali
Den normale dannelse af kraniet sker på grund af tilstedeværelsen af primære vækst- og knogleombygningscentre i det – kraniofaciale synarthroser (led), som lukker sig på et bestemt tidspunkt under udviklingen af hovedskelettet og sikrer knoglernes sammensmeltning. [ 6 ]
Pandebenet (os frontale) i den nyfødtes kranium består af to halvdele, mellem hvilke der er en lodret fiberforbindelse - den mediane pande- eller metopiske sutur (fra græsk metopon - pande), som løber fra toppen af næseryggen op langs pandens midterlinje til den forreste fontanel. Dette er den eneste fiberagtige kraniesutur, der heler i spædbarnsalderen: fra 3-4 måneder til 8-18 måneder. [ 7 ]
Se også – Ændringer i kraniet efter fødslen
Årsagerne til trigonocephali er metopisk kraniosynostose (craniosynostose) eller metopisk synostose (fra græsk syn – sammen og osteon – knogle), det vil sige for tidlig (før den tredje måned) immobil sammenvoksning af knoglerne i kraniehvælvet langs den mediane frontale sutur. Således er kraniosynostose og trigonocephali relateret som årsag og virkning eller som en patologisk proces og dens resultat. [ 8 ]
I de fleste tilfælde er trigonocephali hos et barn resultatet af primær (isoleret) kraniosynostose, hvis nøjagtige årsag er ukendt. Isoleret kraniosynostose forekommer sporadisk, sandsynligvis på grund af en kombination af genetiske og miljømæssige faktorer. [ 9 ]
Men trigonocephali kan være en del af medfødte syndromer, der opstår på grund af kromosomale abnormiteter og mutationer i forskellige gener. Disse omfatter: Opitz trigonocephali syndrom (Bohring-Opitz syndrom), Apert syndrom, Loeys-Dietz syndrom, Pfeiffer syndrom, Jackson-Weiss syndrom, kraniofacial dysostose eller Crouzons syndrom, Jacobsens, Saethre-Chotzens, Münkes syndromer. I sådanne tilfælde kaldes trigonocephali syndromisk. [ 10 ]
Ved fødslen er hjernens størrelse normalt 25 % af dens voksenstørrelse, og ved udgangen af det første leveår når den omkring 75 % af den voksne hjernes størrelse. Men ved primær forsinkelse i hjernens vækst er såkaldt sekundær kraniosynostose mulig. Ætiologien for forsinkelsen er forbundet med metaboliske forstyrrelser, nogle hæmatologiske sygdomme og teratogene virkninger af kemikalier på fosteret (herunder dem i lægemidler). [ 11 ]
Ifølge eksperter fortsætter trigonocephali hos voksne, der ikke blev behandlet i barndommen, som følge af isoleret kraniosynostose eller medfødt syndrom, hele livet. [ 12 ]
Risikofaktorer
Eksperter mener, at de vigtigste risikofaktorer for trigonocephali (og metopisk kraniosynostose som årsag) er genetiske: I løbet af de sidste to årtier er der identificeret mere end 60 gener, hvis mutationer er forbundet med for tidlig, immobil fusion af kranieknoglerne hos spædbørn.
Risikoen for at udvikle patologier som kraniofacial synarthrose og generel osteogenese (knogledannelse) øges i tilfælde af unormal fosterpræsentation, intrauterin hypoxi, flerfoldsgraviditet, alkoholforbrug, stofmisbrug eller rygning under graviditet. [ 13 ]
Patogenese
Ifølge den fremherskende teori er patogenesen af trigonocephali rodfæstet i en forstyrrelse af føtal osteogenese i de tidlige stadier af graviditeten, oftest forårsaget af genetiske faktorer, da tilfældige kromosomafvigelser opdages hos nyfødte med metopisk kraniosynostose. For eksempel er en af de mest almindelige trisomi 9p, som fører til kraniofaciale og skeletale defekter, mental retardering og psykomotorisk udvikling. [ 14 ]
På grund af den for tidlige sammenvoksning af den mediane pande sutur hæmmes væksten i dette område af kraniet: den laterale vækst af pandebenet begrænses af en forkortelse af den forreste kraniefossa; en knogleformet ryg dannes langs pandens midterlinje; der er en konvergens af knoglerne, der danner øjenhulerne, og en fordybning af temporalknoglerne. [ 15 ]
Men kraniets vækst i andre områder fortsætter: der er kompenserende sagittal (for-bag) og tværgående vækst af kraniets bagside (med udvidelse af dens parietal-occipitale del), samt vertikal og sagittal vækst af den øvre del af ansigtet. Som følge af disse lidelser får kraniet en uregelmæssig form - trekantet.
Symptomer trigonocephali
De vigtigste symptomer på trigonocephali er ændringer i hovedets form og udseende:
- set ovenfra hovedets krone har kraniet form som en trekant;
- indsnævret pande;
- en mærkbar eller håndgribelig højderyg (knoglefremspring), der løber langs midten af panden, hvilket giver pandebenet et spids (kølformet) udseende;
- deformation af den øvre del af øjenhulerne (udfladning af de supraorbitale kamme) og hypotelorisme (reduceret afstand mellem øjnene).
Den frontale (anteriore) fontanelle kan også lukke sig for tidligt.
Ved syndromisk trigonocephali er der andre anomalier og tegn på mental retardering hos børn. [ 16 ]
Komplikationer og konsekvenser
Diagnosticering trigonocephali
Trigonocephali diagnosticeres ved fødslen eller inden for få måneder efter fødslen. De mindre alvorlige fund ved metopisk kraniosynostose opdages dog muligvis ikke før tidlig barndom.
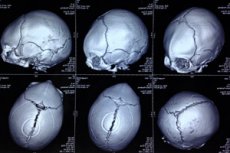
For at visualisere kraniets patologi udføres instrumentel diagnostik ved hjælp af computertomografi af hovedet og ultralyd. [ 19 ], [ 20 ]
Differential diagnose
Differentialdiagnose er nødvendig for at skelne en syndromisk defekt fra isoleret metopisk synostose, hvor barnet får foretaget en genotypetest.
Behandling trigonocephali
Hos nogle børn er tilfældene af metopisk synostose ret milde (når der kun er en synlig rille i panden og ingen andre symptomer), og kræver ikke specifik behandling. [ 21 ]
Behandling af svær trigonocephali er kirurgisk og involverer en operation for at korrigere hovedets form og sikre normal hjernevækst, samt kirurgisk korrektion af ansigtsknogledeformiteter. [ 22 ]
Denne kirurgiske procedure – synostektomi af den metopiske sutur, orbital randforskydning og kranioplastik – udføres før 6-månedersalderen. Barnet overvåges indtil etårsalderen; i løbet af de første leveår undersøges barnet regelmæssigt for at sikre, at der ikke er problemer med tale, motorik eller adfærd. [ 23 ]
Forebyggelse
Der findes ingen metoder til at forhindre denne medfødte defekt, men genetisk rådgivning kan forhindre fødslen af et barn med en uhelbredelig kraniocerebral patologi.
Kraniosynostose hos et foster kan opdages ved at udføre en prænatal ultralyd af dets hoved i andet og tredje trimester af graviditeten.
Vejrudsigt
Prognosen afhænger i høj grad af graden af kraniedeformation, som påvirker hjernens neurokognitive funktioner. Og hvis der ikke er udført korrigerende kirurgi, har børn med trigonocephali – sammenlignet med raske jævnaldrende – lavere generelle kognitive evner, problemer med tale, syn, opmærksomhed og adfærd.
Использованная литература