Medicinsk ekspert af artiklen

Nye publikationer
Martin-Bell-syndromet
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
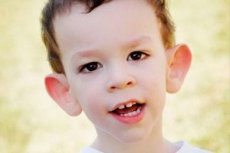
Martin-Bell syndrom blev beskrevet i 1943 af læger, som det blev opkaldt efter. Sygdommen er en genetisk lidelse, der består af mental retardering. I 1969 blev ændringer i kromosom X (skørhed i den distale arm), der er karakteristiske for denne sygdom, identificeret. I 1991 opdagede forskere det gen, der er ansvarligt for udviklingen af denne sygdom. Denne sygdom kaldes også "fragilt X-syndrom". Både drenge og piger er modtagelige for sygdommen, men drenge er oftere (3 gange) ramt.
Epidemiologi
Martin-Bell syndrom er en ret almindelig sygdom: 0,3-1,0 ud af 1.000 mænd lider af denne sygdom, og 0,2-0,6 ud af 1.000 kvinder. Desuden fødes børn med Martin-Bell syndrom på alle kontinenter med samme hyppighed. Nationalitet, hudfarve, øjenform, levevilkår og folks velbefindende påvirker naturligvis ikke sygdommens forekomst. Dens forekomstfrekvens kan kun sammenlignes med hyppigheden af Downs syndrom (1 sygdom ud af 600-800 nyfødte). En femtedel af mandlige bærere af det ændrede gen er raske, har ingen kliniske eller genetiske abnormiteter, resten har tegn på mental retardering fra mild til svær form. Blandt kvindelige bærere er lidt mere end en tredjedel syge.
Fragilt X-syndrom rammer cirka 1 ud af 2.500-4.000 mænd og 1 ud af 7.000-8.000 kvinder. Bærerprævalensen blandt kvinder anslås at være 1 ud af 130-250; bærerprævalensen blandt mænd anslås at være 1 ud af 250-800.
Årsager Martin-Bell-syndromet
Martin-Bell syndrom udvikler sig på grund af fuldstændig eller delvis ophør af kroppens produktion af et specifikt protein. Dette sker på grund af manglende respons fra FMR1-genet, lokaliseret i X-kromosomet. Mutationen sker som følge af genets omstrukturering fra ustabile strukturelle varianter af gentilstandene (alleler), og ikke helt fra begyndelsen. Sygdommen overføres kun gennem den mandlige linje, og manden er ikke nødvendigvis syg. Mandlige bærere overfører genet til deres døtre i uændret form, så deres mentale retardering er ikke tydelig. Ved yderligere overførsel af genet fra moderen til hendes børn muterer genet, og alle de tegn, der er karakteristiske for denne sygdom, fremkommer.
Patogenese
Patogenesen af Martin-Bell syndrom er baseret på mutationer i genapparatet, som fører til blokering af produktionen af FMR-protein, et protein der er afgørende for kroppen, især i neuroner, og som findes i forskellige væv. Forskning viser, at FMR-proteiner er direkte involveret i de processer, der regulerer translationer, der forekommer i hjernevæv. Fraværet af dette protein eller kroppens begrænsede produktion fører til mental retardering.
I sygdommens patogenese betragtes genhypermetylering som en nøglelidelse, men det har endnu ikke været muligt endeligt at identificere mekanismen for udviklingen af denne lidelse.
Samtidig blev patologiens locus heterogenitet også opdaget, hvilket er forbundet med polyallelisme, såvel som polylocus. Tilstedeværelsen af alleliske varianter af sygdomsudviklingen blev bestemt, som er forårsaget af eksistensen af punktmutationer, såvel som ødelæggelsen af FMRL-typegenet.
Patienterne har også 2 fragile triplets, der er følsomme over for folsyre, placeret 300 kb, samt 1,5-2 millioner bp fra den fragile triplet, der indeholder FMR1-genet. Mekanismen for mutationer, der forekommer i FRAXE- og FRAXF-generne (de er identificeret i de ovennævnte fragile triplets) er relateret til mekanismen for lidelser i Martin Bell syndrom. Denne mekanisme er forårsaget af spredningen af GCC- og CGG-gentagelser, som forårsager methylering af de såkaldte CpG-øer. Ud over den klassiske form af patologien er der også 2 sjældne typer, der adskiller sig på grund af ekspansionen af trinukleotidgentagelser (i mandlig og kvindelig meiose).
Det blev konstateret, at patienten i den klassiske form af syndromet mangler et særligt nukleocytoplasmatisk protein af FMR1-typen, som udfører funktionen med at binde forskellige mRNA'er. Derudover fremmer dette protein dannelsen af et kompleks, der hjælper med at udføre translationsprocesser inde i ribosomer.
Symptomer Martin-Bell-syndromet
Hvordan genkender man sygdommen hos børn? Hvad er de første tegn? I de første måneder af et barns liv er det umuligt at genkende Martin-Bell-symptomet, bortset fra at der nogle gange observeres et fald i muskeltonus. Efter et år er det kliniske billede af sygdommen mere tydeligt: barnet begynder at gå og tale sent, nogle gange er tale helt fraværende. Han er hyperaktiv, vifter tilfældigt med armene, er bange for folkemængder og støj, er stædig, der er skarpe vredesudbrud, følelsesmæssig ustabilitet, epileptiske anfald forekommer, får ikke øjenkontakt. Hos patienter med Martin-Bell syndrom afsløres sygdommen også af deres udseende: ørerne er udstående og store, panden er tung, ansigtet er aflangt, hagen er udstående, strabismus, brede hænder og fødder. De er også karakteriseret ved endokrine lidelser: ofte stor vægt, fedme, store testikler hos mænd, tidlig pubertet.

Blandt patienter med Martin-Bell syndrom varierer intelligensniveauet meget: fra mild mental retardering til alvorlige tilfælde. Hvis en normal person har en intelligenskvotient (IQ) på 100 i gennemsnit, og et geni har 130, så har personer, der er modtagelige for sygdommen, 35-70.
Alle kliniske symptomer på patologien kan karakteriseres ved en triade af hovedmanifestationer:
- oligofreni (IQ er 35-50);
- dysmorfofobi (observeres fremspringende ører og prognatisme);
- makroorchidisme, som opstår efter pubertetens begyndelse.
Cirka 80% af patienterne har også bikuspidalklapprolaps.
Syndromets fulde form manifesterer sig dog kun hos 60% af alle patienter. Hos 10% opdages kun mental retardering, og hos resten udvikler sygdommen sig med en anden kombination af symptomer.
Blandt de første tegn på sygdommen, som optræder i en tidlig alder:
- det syge barn udviser betydelig mental retardering i sammenligning med andre jævnaldrendes udvikling;
- opmærksomheds- og koncentrationsforstyrrelser;
- stærk stædighed;
- børn begynder at gå og tale ret sent;
- hyperaktivitet og taleudviklingsforstyrrelser observeres;
- meget stærke og ukontrollerbare vredesanfald;
- mutisme kan udvikle sig - dette er en fuldstændig mangel på tale hos et barn;
- barnet oplever social angst og er i stand til at gå i panik på grund af høj støj eller andre høje lyde;
- barnet vifter ukontrolleret og kaotisk med armene;
- generthed observeres, barnet er bange for at være på overfyldte steder;
- fremkomsten af forskellige obsessive ideer, ustabil følelsesmæssig tilstand;
- Barnet kan være tilbageholdende med at få øjenkontakt med mennesker.
Hos voksne observeres følgende symptomer på patologien:
- specifikt udseende: et aflangt ansigt med en tung pande, store fremstående ører, en stærkt fremstående hage;
- platfod, mellemørebetændelse og strabismus;
- puberteten indtræffer ret tidligt;
- fedme kan udvikle sig;
- Ofte observeres hjertefejl ved Martin-Bell syndrom;
- hos mænd observeres forstørrelse af testiklerne;
- leddenes artikulationer bliver meget mobile;
- vægt og højde stiger kraftigt.
Diagnosticering Martin-Bell-syndromet
For at diagnosticere Martin Bell syndrom skal du kontakte en kvalificeret genetiker. Diagnosen stilles efter specifikke genetiske tests, der giver dig mulighed for at identificere det defekte kromosom.
Test
I et tidligt stadie af sygdommen anvendes en cytogenetisk metode, hvor et fragment af cellemateriale tages fra patienten, hvortil folinsyre tilsættes for at fremkalde ændringer i kromosomerne. Efter en vis periode identificeres et område af kromosomet, hvor der er en mærkbar udtynding - dette er et tegn på tilstedeværelsen af fragilt X-syndrom.
Denne test er dog ikke egnet til diagnose i de senere stadier af sygdommen, fordi dens nøjagtighed reduceres af den udbredte brug af multivitaminer, der indeholder folsyre.
Integreret diagnostik af Martin-Bell syndrom er en molekylærgenetisk undersøgelse, som består i at bestemme antallet af såkaldte trinukleotidgentagelser i genet.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentel diagnostik
En meget specifik metode til instrumentel diagnostik er PCR (polymerasekædereaktion), som gør det muligt at studere strukturen af aminosyrerester indeholdt i X-kromosomet og derved bestemme tilstedeværelsen af Martin Bell syndrom.
Der findes også en separat, endnu mere specifik metode til patologidiagnostik – en kombination af PCR og detektion ved hjælp af kapillærelektroforese. Denne metode er yderst nøjagtig og detekterer kromosomal patologi hos patienter med primær ovariesvigt samt ataksisk syndrom.
Tilstedeværelsen af defekten kan bestemmes efter udførelse af diagnostik på EEG. Patienter med denne sygdom har lignende bioelektrisk hjerneaktivitet.
Hvilke tests er nødvendige?
Differential diagnose
Differentierede metoder, der hjælper med at mistænke syndromet, omfatter:
- klinisk - 97,5% af patienterne har tydelige tegn på mental retardering (moderat eller alvorlig); 62% har store, fremstående ører; 68,4% har en stor, fremstående hage og pande; 68,4% af drengene har forstørrede testikler, 41,4% har talevanskeligheder (ujævn talehastighed, ukontrollerbar lydstyrke osv.);
- cytogen - blod undersøges for lymfocytkultur, antallet af celler med skrøbeligt X-kromosom pr. 100 undersøgte celler bestemmes;
- Elektroencefalografi - ændringer i hjernens elektriske impulser, der er specifikke for Martin-Bell syndrom, registreres.
Hvem skal kontakte?
Behandling Martin-Bell-syndromet
Ved behandling af voksne patienter anvendes antidepressiva med psykostimulerende midler. Lægemiddelbehandlingsprocessen overvåges konstant af en psykolog og psykiater. Derudover udføres mikroinjektionsprocedurer med lægemidler som Cerebrolysin (eller dets derivater) samt cytomediciner (såsom Solcoseryl eller Lidase) i private klinikker.
Ved udvikling af ataksisk syndrom anvendes blodfortyndende lægemidler og nootropika. Derudover ordineres aminosyreblandinger og angioprotektorer. Kvinder med primær ovariesvigt ordineres korrigerende behandling med naturlægemidler og østrogener.
Glutaminreceptorantagonister anvendes også i behandlingen.
Traditionelt involverer behandlingen af Martin-Bell syndrom brug af medicin, der påvirker sygdommens symptomer, men ikke dens årsag. Denne terapi involverer ordination af antidepressiva, neuroleptika og psykostimulerende midler. Ikke alle lægemidler er indiceret til brug hos børn, så listen over lægemidler er ret begrænset. Neuroleptika, der kan anvendes efter 3 år (den tidligste alder for deres ordination), omfatter haloperidol i dråber og tabletter, chlorpromazin i opløsning og periciazin i dråber. Således beregnes dosis af haloperidol til børn afhængigt af kropsvægt. For voksne ordineres dosis individuelt. Det tages oralt, startende med 0,5-5 mg 2-3 gange dagligt, hvorefter dosis gradvist øges til 10-15 mg. Når der opstår forbedring, skiftes der til en lavere dosis for at opretholde den opnåede tilstand. Ved psykomotorisk agitation ordineres 5-10 mg intramuskulært eller intravenøst, flere gentagelser er mulige efter 30-40 minutter. Den daglige dosis bør ikke overstige 100 mg. Bivirkninger i form af kvalme, opkastning, muskelspasmer, øget tryk, arytmi osv. er mulige. Ældre bør tage særlige forholdsregler, da der er registreret tilfælde af pludseligt hjertestop, og tardiv dyskinesi (ufrivillige bevægelser) kan forekomme.
Antidepressiva øger aktiviteten i hjernestrukturer, lindrer depression, spændinger og forbedrer humøret. Disse lægemidler, der anbefales til brug fra 5-8 år ved Martin-Bell syndrom, omfatter clomipromin, sertralin, fluoxetin og fluvoxamin. Fluoxetin tages derfor oralt under måltider 1-2 gange (helst i første halvdel af dagen), startende med 20 mg om dagen, øget til 80 mg om nødvendigt. Ældre personer anbefales ikke en dosis højere end 60 mg. Behandlingsforløbet bestemmes af en læge, men ikke mere end 5 uger.
Mulige bivirkninger: svimmelhed, angst, tinnitus, appetitløshed, takykardi, ødem osv. Forsigtighed er påkrævet ved ordination til ældre, personer med hjerte-kar-sygdomme og diabetes.
Psykostimulanter er psykotrope lægemidler, der bruges til at forbedre opfattelsen af eksterne stimuli: de skærper hørelse, reaktioner og syn.
Diazepam ordineres som et beroligende middel mod neuroser, angst, epileptiske anfald og kramper. Det tages oralt, intravenøst, intramuskulært, rektalt (i endetarmen). Det ordineres individuelt, afhængigt af sygdommens sværhedsgrad, med de mindste doser på 5-10 mg, dagligt - 5-20 mg. Behandlingsvarigheden er 2-3 måneder. For børn beregnes dosis under hensyntagen til kropsvægt og individuelle karakteristika. Bivirkninger omfatter sløvhed, apati, døsighed, kvalme, forstoppelse. Det er farligt at kombinere med alkohol, da afhængighed af lægemidlet er mulig.
Ved behandling af Martin-Bell syndrom har der været tilfælde af forbedring af tilstanden og med introduktionen af lægemidler fremstillet af animalsk materiale (hjerne): cerebrolysat, cerebrolysin, cerebrolysat-M. Hovedkomponenterne i disse lægemidler er peptider, der fremmer produktionen af protein i neuroner og dermed genopfylder det manglende protein. Cerebrolysin administreres som en stråle på 5-10 ml, behandlingsforløbet består af 20-30 injektioner. Lægemidlet ordineres til børn fra et år, administreres intramuskulært hver dag 1-2 ml i en måned. Gentagne administrationssessioner er mulige. Bivirkninger i form af feber, kontraindiceret for gravide kvinder.
Der blev forsøgt at behandle sygdommen med folsyre, men kun det adfærdsmæssige aspekt blev forbedret (niveauet af aggression og hyperaktivitet faldt, tale forbedret), og intet ændrede sig på det intellektuelle niveau. For at forbedre sygdommens tilstand ordineres folsyre, fysioterapimetoder, taleterapi, pædagogisk og social korrektion er indiceret.
Litiumpræparater anses også for at være effektive, da de hjælper med at forbedre patientens tilpasning til det sociale miljø, såvel som kognitiv aktivitet. Derudover regulerer de også hans adfærd i samfundet.
Brug af urter til Martin-Bell syndrom er mulig som antidepressiva. Urter, der hjælper med at lindre spændinger, angst og forbedre søvn, omfatter baldrian, pebermynte, timian, perikon og kamille. Infusioner tilberedes som følger: til 1 teskefuld tørrede urter skal du bruge et glas kogende vand, afkog trækkes i mindst 20 minutter, tages hovedsageligt om aftenen før sengetid eller om eftermiddagen. En skefuld honning ville være et godt supplement til dem.
Fysioterapibehandling
For at eliminere neurologiske manifestationer udføres særlige fysioterapeutiske procedurer - såsom øvelser i poolen, muskelafslapning og akupunktur.
Kirurgisk behandling
Et vigtigt trin i behandlingen anses også for at være plastikkirurgiske metoder - operationer, der hjælper med at forbedre patientens udseende. Plastikkirurgi udføres på lemmer og ører, såvel som kønsorganerne. Korrektion af gynækomasti med epispadi udføres også, såvel som andre defekter i udseende.
Forebyggelse
Den eneste metode til at forebygge sygdommen er prænatal screening af gravide kvinder. Der findes særlige undersøgelser, der muliggør tidlig opdagelse af patologien, hvorefter det anbefales at afbryde graviditeten. Alternativt anvendes IVF, som kan hjælpe barnet med at arve et sundt X-kromosom.
Forebyggelse af patienten afhænger af, om genmutationen er opstået igen eller er blevet arvet. Til dette formål udføres molekylærgenetisk diagnostik. Det faktum, at testen ikke afslørede det "skrøbelige X-kromosom" hos slægtninge, taler for mutationens "friskhed", hvilket betyder, at risikoen for at få et barn med Martin-Bell syndrom er meget lille. I familier med syge mennesker vil testen hjælpe med at undgå gentagne tilfælde.
Vejrudsigt
Prognosen for Martin-Bell syndrom er gunstig for livet, men ikke for helbredelse. Den forventede levetid afhænger af sygdommens sværhedsgrad og tilhørende defekter. Patienten kan leve et normalt liv. Ved alvorlige former for Martin-Bell syndrom er patienterne i risiko for livslang invaliditet.
Forventet levetid
Martin Bell syndrom har ikke en alvorlig negativ indvirkning på helbredet, så den forventede levetid for de fleste mennesker, der er blevet diagnosticeret med denne patologi, adskiller sig ikke fra standardindikatorer.