Medicinsk ekspert af artiklen

Nye publikationer
Huntingtons sygdom
Sidst revideret: 05.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
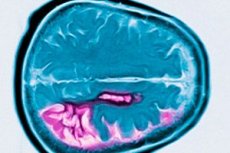
Huntingtons sygdom er en autosomal dominant neurodegenerativ lidelse karakteriseret ved progressiv kognitiv tilbagegang, ufrivillige bevægelser og nedsat motorisk koordination, der begynder i middelalderen. Diagnosen bekræftes ved genetisk testning. Behandlingen er primært symptomatisk. Genetisk testning kan anbefales til blodsbeslægtede. George Huntington beskrev først tilstanden i 1872 efter at have studeret et familietilfælde blandt beboere på Long Island.
Prævalensen af Huntingtons sygdom er cirka 10 tilfælde pr. 100.000 indbyggere, og på grund af dens sene debut har cirka 30 personer ud af 100.000 en 50% risiko for at udvikle den i deres levetid. Selvom sygdommen oftest optræder mellem 35 og 40 år, er aldersintervallet for debut ret bredt, med den tidligste debut i 3-årsalderen og den seneste i 90-årsalderen. Selvom sygdommen oprindeligt blev antaget at have 100% penetrans, menes dette nu ikke altid at være tilfældet. Hos personer, der arvede genet for sygdommen fra deres far, manifesterer sygdommen sig i gennemsnit 3 år tidligere end hos dem, der arvede det patologiske gen fra deres mor. Hos cirka 80% af patienterne, der arvede det patologiske gen fra deres far, manifesterer sygdommen sig før 20-årsalderen. Fænomenet med tidligere manifestation af en genetisk defekt hos afkom kaldes anticipation.
 [ 1 ]
[ 1 ]
Hvad forårsager Huntingtons sygdom?
Huntingtons sygdom har ingen kønspræference. Der påvises atrofi af nucleus caudatus, hvor små neuroner degenererer, og niveauet af neurotransmittere - gamma-aminosmørsyre (GABA) og substans P - falder.
Et mutantgen med et øget antal ("ekspansion") af CAG (cystein-alanin-glycin) DNA-sekvenser, der koder for aminosyren glutamin, er ansvarlig for udviklingen af Huntingtons sygdom. Produktet af dette gen, det store protein huntingtin, indeholder en for stor mængde polyglutaminrester, hvilket fører til sygdommen via en ukendt mekanisme. Jo flere CAG-gentagelser, desto tidligere debuterer sygdommen, og desto mere alvorligt er dens forløb. Fra generation til generation kan antallet af gentagelser stige, hvilket over tid fører til en forværring af familiens fænotype.
Trods betydelig interesse for de genetiske og biokemiske ændringer i Parkinsons sygdom, var søgen efter et gen for sygdommen forgæves indtil slutningen af 1970'erne. På det tidspunkt organiserede Nancy Wexler og Allan Tobin en workshop sponsoreret af Hereditary Disease Foundation for at diskutere en strategi til at finde et gen for Huntingtons sygdom. David Housman, David Botstein og Ray White, der deltog i mødet, foreslog, at nyligt udviklede rekombinante DNA-teknikker kunne hjælpe med at nå dette mål. En central opgave i projektet var at finde en stor familie med mange generationer af Huntingtons sygdom for at få DNA-prøver. I 1979 blev et fælles projekt mellem forskere fra Venezuela og USA lanceret for at undersøge en stor familie med Huntingtons sygdom, der boede ved bredden af Maracheibo-søen (Venezuela). I 1983 blev Huntingtons sygdomsgenet lokaliseret i enden af den korte arm på kromosom 4 (Gusella et al., 1983), og et årti senere blev det afsløret, at mutationen af dette gen består af en stigning i antallet af gentagelser af cytosin-adenin-guanin (CAG) trinukleotidet (Huntington's Disease Collaborative Research Group, 1993). Den metode, der er udviklet af denne videnskabelige gruppe, betragtes i øjeblikket som standard for positionskloning af nye gener.
Mens vildtypegenet har en strækning på 10-28 CAG-gentagelser, har den mutante form af genet, der forårsager Huntingtons sygdom, en øget strækning fra 39 til mere end 100 CAG-gentagelser. Opdagelsen af udvidelsen af trinukleotidgentagelser har bidraget til at forklare mange af sygdommens kliniske træk. Især blev der fundet en omvendt korrelation mellem debutalderen og længden af regionen med gentagne trinukleotider. Forventning om faderlig arv kan forklares ved, at en stigning i antallet af gentagelser ofte forekommer hos mænd under spermatogenesen. Analyse af nye mutationer har vist, at de normalt forekommer, når en af forældrene, normalt faderen, havde et CAG-gentagelsestal højere end 28; i dette tilfælde steg antallet af disse gentagelser i den næste generation. Det er nu blevet fastslået, at hvis antallet af gentagelser ikke er mere end 28, overføres det stabilt fra generation til generation. Hvis antallet af gentagelser er fra 29 til 35, opstår der ingen symptomer på Huntingtons sygdom, men når den overføres til afkommet, kan længden af denne region øges. Hvis antallet af gentagelser er fra 36 til 39, kan sygdommen i nogle tilfælde (men ikke altid) manifestere sig klinisk (ufuldstændig penetrans), og når den overføres til afkommet, er en stigning i antallet af trinukleotidgentagelser mulig. Hvis antallet af gentagelser overstiger 40, forekommer sygdommen i næsten alle tilfælde, og når den overføres til afkommet, er yderligere udvidelse af gentagelser mulig. Årsagerne til stigningen i antallet af gentagelser forbliver ukendte.
Patomorfologi af Huntingtons sygdom
Huntingtons sygdom er karakteriseret ved neuronalt tab, overvejende i nucleus caudatus og putamen, og til en vis grad også i cortex og andre hjernestrukturer. Den samlede hjernevægt ved Huntingtons sygdom reduceres ikke kun af et fald i antallet af neuroner, men også af tabet af hvid substans. I hjernebarken er celler i lag V og VI mest påvirket. Sværhedsgraden af mikro- og makroskopiske degenerative forandringer (justeret for alder ved død) korrelerer med antallet af CAG-gentagelser. Detaljeret patologisk analyse af forandringer i flere hundrede tilfælde af Huntingtons sygdom har vist, at degeneration af striatum begynder i den dorsomediale del af nucleus caudatus og den dorsolaterale del af putamen, og derefter spredes ventralt. Forskellige grupper af neuroner i nucleus caudatus og putamen påvirkes i forskellig grad. Interneuroner i striatum forbliver relativt intakte, men nogle projektionsneuroner påvirkes selektivt. I den juvenile form af Huntingtons sygdom er patomorfologiske ændringer i striatum mere udtalte og mere udbredte og involverer hjernebarken, lillehjernen, thalamus og globus pallidus.
Neurokemiske ændringer ved Huntingtons sygdom
GABA. Neurokemiske undersøgelser af hjernen hos patienter med Huntingtons sygdom afslørede et signifikant fald i GABA-koncentrationen i striatum. Efterfølgende undersøgelser bekræftede, at Huntingtons sygdom er forbundet med et fald i antallet af GABAerge neuroner og viste, at GABA-koncentrationerne er reduceret ikke kun i striatum, men også i dets projektionszoner - de eksterne og interne segmenter af globus pallidus og substantia nigra. I hjernen ved Huntingtons sygdom blev ændringer i GABA-receptorer også påvist ved hjælp af receptorbindingsstudier og in situ-hybridisering af mRNA. Antallet af GABA-receptorer var moderat reduceret i nucleus caudatus og putamen, men steget i den retikulære del af substantia nigra og det eksterne segment af globus pallidus, hvilket sandsynligvis skyldes denervationshypersensitivitet.
Acetylcholin. Acetylcholin bruges som neurotransmitter af store ikke-tornede interneuroner i striatum. Tidlige post mortem-studier af patienter med Huntingtons sygdom viste nedsat kolinacetyltransferase (ChAT)-aktivitet i striatum, hvilket tyder på et tab af kolinerge neuroner. Sammenlignet med den signifikante reduktion i GABAerge neuroner er kolinerge interneuroner imidlertid relativt skånet. Derfor er tætheden af acetylcholinesterase-positive neuroner og ChAT-aktivitet i striatum faktisk relativt forhøjet sammenlignet med aldersmatchede kontrolpersoner.
Substans P. Substans P findes i mange mellemstore, tornede neuroner i striatum, som overvejende projicerer til det indre segment af globus pallidus og substantia nigra, og som normalt også indeholder dynorphin og GABA. Substans P-niveauerne i striatum og pars reticularis af substantia nigra er reducerede ved Huntingtons sygdom. I sygdommens terminale stadium har immunhistokemiske undersøgelser vist en signifikant reduktion i antallet af neuroner, der indeholder substans P. I tidligere stadier er neuroner, der indeholder substans P og projicerer til det indre segment af globus pallidus, relativt skånede sammenlignet med neuroner, der projicerer til pars reticularis af substantia nigra.
Opioidpeptider. Enkephalin findes i de mellemstore, tornede projektioner, GABAerge neuroner i den indirekte signalvej, som projicerer til det eksterne segment af globus pallidus og bærer D2-receptorer. Immunhistokemiske undersøgelser har vist, at enkephalinholdige neuroner, der projicerer til det eksterne segment af globus pallidus, går tabt tidligt i Huntingtons sygdom. Disse celler dør tilsyneladende tidligere end substans P-holdige celler, der projicerer til det interne segment af globus pallidus.
Katekolaminer. Neuroner, der indeholder biogene aminer (dopamin, serotonin), og som projicerer til striatum, er placeret i den kompakte del af substantia nigra, ventral tegmentum og raphe-kernerne. Mens noradrenerge projektioner til det humane striatum er minimale, er serotonin- og dopaminniveauerne (pr. gram væv) i striatum forhøjede, hvilket indikerer bevarelsen af disse afferente projektioner på trods af det markante tab af striatums egne neuroner. Dopaminerge neuroner i substantia nigra forbliver intakte i både klassiske og juvenile former for Huntingtons sygdom.
Somatostatin/neuropeptid Y og nitrogenoxidsyntetase. Måling af somatostatin- og neuropeptid Y-niveauer i striatum ved Huntingtons sygdom viste en 4-5 gange stigning sammenlignet med normalt væv. Immunhistokemiske undersøgelser viste absolut bevarelse af striatale interneuroner indeholdende neuropeptid Y, somatostatin og nitrogenoxidsyntetase. Disse neuroner er således resistente over for den patologiske proces.
Excitatoriske aminosyrer. Det er blevet foreslået, at selektiv celledød ved Huntingtons sygdom skyldes en glutamatinduceret neurotoksisk effekt. Niveauerne af glutamat og quinolinsyre (et endogent neurotoksin, der er et biprodukt af serotoninmetabolisme og en agonist af glutamatreceptorer) i striatum ved Huntingtons sygdom er en smule ændrede, men en nylig undersøgelse med MR-spektroskopi afslørede en stigning i glutamatniveauer in vivo. Niveauet af det gliale enzym, der er ansvarligt for syntesen af quinolinsyre i striatum ved Huntingtons sygdom, er forhøjet med ca. 5 gange sammenlignet med normalt, mens aktiviteten af det enzym, der sikrer nedbrydningen af quinolinsyre, kun er forhøjet ved Huntingtons sygdom med 20-50%. Syntesen af quinolinsyre kan således være forhøjet ved Huntingtons sygdom.
Studier af excitatoriske aminosyrereceptorer (EAA) ved Huntingtons sygdom har vist en signifikant reduktion i antallet af NMDA-, AMPA-, kainat- og metabotrope glutamatreceptorer i striatum, samt AMPA- og kainatreceptorer i hjernebarken. I det sene stadie af Huntingtons sygdom var NMDA-receptorer stort set fraværende, mens der i de prækliniske og tidlige stadier blev observeret en signifikant reduktion i antallet af disse receptorer.
Selektiv følsomhed. Ved Huntingtons sygdom går visse typer striatale celler selektivt tabt. De mellemstore, tornede neuroner, der projicerer til det eksterne segment af globus pallidus og indeholder GABA og enkephalin, dør meget tidligt i sygdommen, ligesom de neuroner, der indeholder GABA og substans P og projicerer til den retikulære del af substantia nigra. Tabet af neuroner, der indeholder GABA og enkephalin og projicerer til det eksterne segment af globus pallidus, desinficerer denne struktur, hvilket igen fører til aktiv hæmning af nucleus subthalamus. Den nedsatte aktivitet i nucleus subthalamus kan tilsyneladende forklare de choreiforme bevægelser, der forekommer ved Huntingtons sygdom. Det har længe været kendt, at fokale læsioner i nucleus subthalamus kan forårsage chorea. Tab af GABA- og substans P-neuroner, der projicerer til substantia nigra pars reticularis, er sandsynligvis ansvarlig for de okulomotoriske forstyrrelser, der ses ved Huntingtons sygdom. Denne signalvej hæmmer normalt substantia nigra pars reticularis-neuroner, der projicerer til den superior colliculus, som igen regulerer saccader. Ved juvenil Huntingtons sygdom er de ovennævnte signalveje mere alvorligt påvirket, og derudover går striatale fremspring til det interne segment af globus pallidus tabt tidligt.
Proteinet huntingtin, kodet af det gen, hvis mutation forårsager Huntingtons sygdom, findes i forskellige strukturer i hjernen og andre væv. Huntingtin findes normalt overvejende i neuroners cytoplasma. Proteinet findes i de fleste neuroner i hjernen, men nyere data viser, at dets indhold er højere i matrixneuroner end i striosomale neuroner og højere i projektionsneuroner end i interneuroner. Således korrelerer neuronernes selektive følsomhed med deres huntingtinindhold, som normalt er til stede i visse neuronale populationer.
Ligesom i hjernen hos patienter med Huntingtons sygdom danner huntingtin tætte aggregater i neuronkernerne hos mus, der er transgene for det N-terminale fragment af Huntingtons sygdomsgenet med et udvidet antal gentagelser. Disse intranukleære inklusioner dannes i striatale projektionsneuroner (men ikke i interneuroner). Hos transgene mus dannes inklusionerne flere uger før symptomernes begyndelse. Disse data tyder på, at huntingtin-protein, der indeholder et øget antal glutaminrester, hvis inklusioner koder for trinukleotidgentagelser, eller et fragment af det, akkumuleres i kernen og følgelig kan forringe dets kontrol af cellulære funktioner.
Symptomer på Huntingtons sygdom
Det er vanskeligt at fastslå præcist, hvornår de første symptomer opstod hos patienter med Huntingtons sygdom, da sygdommen manifesterer sig gradvist. Ændringer i personlighed og adfærd, milde koordinationsforstyrrelser kan forekomme mange år før de mere tydelige symptomer viser sig. Når diagnosen stilles, har de fleste patienter koreiske bevægelser, nedsat koordination af fine bevægelser og langsom generering af frivillige sakkader. Efterhånden som sygdommen skrider frem, forringes evnen til at organisere sine aktiviteter, hukommelsen falder, tale bliver vanskelig, okulomotoriske forstyrrelser og nedsat udførelse af koordinerede bevægelser øges. Selvom der i sygdommens tidlige stadie ikke er ændringer i muskler og kropsholdning, kan der, efterhånden som den skrider frem, udvikles dystoniske kropsholdninger, som over tid kan blive et dominerende symptom. I et sent stadie bliver talen sløret, det bliver betydeligt vanskeligt at synke, og det bliver umuligt at gå. Huntingtons sygdom skrider normalt frem over 15-20 år. I det terminale stadie er patienten hjælpeløs og kræver konstant pleje. Det dødelige udfald er ikke direkte relateret til den primære sygdom, men til dens komplikationer, for eksempel lungebetændelse.
Demens ved Huntingtons sygdom
ICD-10-kode
P02.2. Demens ved Huntingtons sygdom (G10).
Demens udvikler sig som en af manifestationerne af en systemisk degenerativ-atrofisk proces med overvejende skade på hjernens striatale system og andre subcoekale kerner. Den nedarves autosomalt dominant.
Som regel manifesterer sygdommen sig i det tredje eller fjerde årti af livet med koreoform hyperkinese (især i ansigt, arme, skuldre, gang), personlighedsændringer (ophidsede, hysteriske og skizoide typer personlighedsafvigelser), psykotiske lidelser (særlig depression med dysterhed, muthed, dysfori; paranoid stemning).
Af særlig betydning for diagnostikken er kombinationen af koreoform hyperkinese, demens og arvelig byrde. Følgende er specifikt for denne demens:
- langsom progression (i gennemsnit 10-15 år): dissociation mellem den resterende evne til at drage omsorg for sig selv og åbenlys intellektuel inkompetence i situationer, der kræver produktivt mentalt arbejde (konceptuel tænkning, læring af nye ting);
- udtalt ujævnhed i mental præstation, som er baseret på grove opmærksomhedsforstyrrelser og ustabile holdninger ("rykkende" tænkning, svarende til hyperkinesi);
- atypisk karakter af åbenlyse krænkelser af højere kortikale funktioner;
- omvendt sammenhæng mellem stigningen i demens og sværhedsgraden af psykotiske lidelser.
I betragtning af den høje andel af psykotiske (paranoide vrangforestillinger om jalousi, forfølgelse) og dysforiske lidelser i sygdommens kliniske billede, udføres behandlingen ved hjælp af forskellige neuroleptika, der blokerer dopaminerge receptorer (phenothiazin- og butyrophenonderivater) eller reducerer niveauet af dopamin i væv (reserpin).
Haloperidol (2-20 mg/dag), tiaprid (100-600 mg/dag) i højst tre måneder, thioridazin (op til 100 mg/dag), reserpin (0,25-2 mg/dag) og det antikonvulsive lægemiddel clonazepam (1,5-6 mg/dag) anvendes. Disse lægemidler hjælper med at reducere hyperkinese, udglatte affektive spændinger og kompensere for personlighedsforstyrrelser.
Indlæggelsesbehandling af psykiske lidelser udføres under hensyntagen til patientens ledende syndrom, alder og generelle tilstand. Ved ambulant behandling er terapiprincipperne de samme (kontinuerlig vedligeholdelsesbehandling af bevægelsesforstyrrelser, periodisk skift af lægemiddel). Lavere doser af neuroleptika anvendes ved ambulant behandling.
Rehabiliteringsforanstaltninger for mild og moderat demens omfatter ergoterapi, psykoterapi og kognitiv træning. Det er nødvendigt at samarbejde med familiemedlemmer og yde psykologisk støtte til de personer, der passer patienten. Den primære metode til sygdomsforebyggelse er medicinsk og genetisk rådgivning af patientens nærmeste slægtninge med en henvisning til DNA-analyse, når der træffes beslutning om at få børn.
Prognosen er generelt ugunstig. Sygdomsforløbet er langsomt fremadskridende, og sygdommen fører normalt til døden efter 10-15 år.
[ 18 ]
Hvad generer dig?
Behandling af Huntingtons sygdom
Behandling af Huntingtons sygdom er symptomatisk. Chorea og agitation kan delvist undertrykkes med neuroleptika (f.eks. chlorpromazin 25-300 mg oralt 3 gange dagligt, haloperidol 5-45 mg oralt 2 gange dagligt) eller reserpin 0,1 mg oralt én gang dagligt. Dosis øges til det maksimalt tolererede (før bivirkninger opstår, såsom døsighed, parkinsonisme; for reserpin, hypotension). Målet med empirisk behandling er at reducere glutamaterg transmission via Nmethyl-O-aspartatreceptorer og opretholde energiproduktionen i mitokondrierne. Behandling, der sigter mod at øge GABA i hjernen, er ineffektiv.
Genetisk testning og rådgivning er vigtige, fordi symptomerne på sygdommen viser sig efter den fødedygtige alder. Personer med en positiv familiehistorie og dem, der er interesserede i testning, henvises til specialiserede centre under hensyntagen til alle etiske og psykologiske implikationer.
Symptomatisk behandling af Huntingtons sygdom
Der findes ingen effektiv behandling, der kan stoppe udviklingen af Huntingtons sygdom. Der er udført adskillige forsøg med forskellige lægemidler, men der er ikke opnået nogen signifikant effekt. Neuroleptika og andre dopaminreceptorantagonister anvendes i vid udstrækning til at korrigere psykiske lidelser og ufrivillige bevægelser hos patienter med Huntingtons sygdom. Ufrivillige bevægelser afspejler en ubalance mellem det dopaminerge og GABAerge system. Derfor anvendes neuroleptika til at reducere overskydende dopaminerg aktivitet. Disse lægemidler kan dog i sig selv forårsage betydelige kognitive og ekstrapyramidale bivirkninger. Derudover er deres effektivitet ikke bevist, undtagen i tilfælde hvor patienten udvikler psykose eller agitation. Neuroleptika forårsager eller forværrer ofte dysfagi eller andre bevægelsesforstyrrelser. Nyere generationer af neuroleptika, såsom risperidon, clozapin og olanzapin, kan være særligt nyttige i behandlingen af Huntingtons sygdom, fordi de forårsager færre ekstrapyramidale bivirkninger, men kan reducere paranoide symptomer eller øget irritabilitet.
Tetrabenazin og reserpin reducerer også aktiviteten af det dopaminerge system og kan reducere sværhedsgraden af ufrivillige bevægelser i sygdommens tidlige stadier. Disse lægemidler kan dog forårsage depression. Da selve sygdommen ofte forårsager depression, begrænser denne bivirkning brugen af reserpin og tetrabenazin betydeligt. I sygdommens sene stadier dør celler, der bærer dopaminreceptorer, så effektiviteten af dopaminreceptorantagonister svækkes eller går tabt.
Neuroleptika, antidepressiva og angstdæmpende midler bruges til at behandle psykose, depression og irritabilitet hos patienter med Huntingtons sygdom, men de bør kun ordineres, så længe patienten rent faktisk har disse symptomer. Lægemidler, der kan være nyttige på et tidspunkt i sygdommen, kan blive ineffektive eller endda skadelige, efterhånden som sygdommen skrider frem.
GABA-receptoragonister er blevet testet hos patienter med Huntingtons sygdom, da Huntingtons sygdom har vist sig at have et signifikant fald i GABA-niveauer i striatum, samt overfølsomhed over for GABA-receptorer i dets projektionsområder. Benzodiazepiner har vist sig effektive i tilfælde, hvor ufrivillige bevægelser og kognitiv svækkelse forværres af stress og angst. Lave doser af disse lægemidler bør ordineres for at undgå uønskede beroligende virkninger. Hos de fleste patienter med Huntingtons sygdom fører ingen af lægemidlerne til en signifikant forbedring af livskvaliteten.
Ved tidlig debut af Huntingtons sygdom med parkinsonsymptomer kan dopaminerge midler forsøges, men deres effektivitet er begrænset. Desuden kan levodopa forårsage eller øge myoklonus hos disse patienter. Samtidig kan baclofen reducere rigiditet hos nogle patienter med Huntingtons sygdom.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Forebyggende (neuroprotektiv) behandling af Huntingtons sygdom
Selvom den genetiske defekt i Huntingtons sygdom er kendt, er det stadig uklart, hvordan den fører til selektiv neuronal degeneration. Det antages, at forebyggende behandlinger, der sigter mod at reducere oxidativ stress og excitotoksicitet, potentielt kan forsinke eller stoppe sygdomsprogressionen. Situationen kan være noget lignende hepatolentikulær degeneration, hvor den genetiske defekt forblev ukendt i mange år, men forebyggende behandlinger rettet mod den sekundære effekt, kobberophobning, blev "kureret". I denne henseende har hypotesen om, at Huntingtons sygdom er forbundet med en forstyrrelse i energimetabolismen og celledød på grund af excitotoksicitet, tiltrukket sig særlig opmærksomhed. Selve sygdommen kan forårsage celledød på grund af intranukleær aggregering af N-terminale fragmenter af huntingtin, hvilket forstyrrer cellulære og metaboliske funktioner. Denne proces kan påvirke nogle grupper af neuroner i større grad end andre på grund af deres højere følsomhed over for excitotoksisk skade. I dette tilfælde vil forebyggende behandling med excitatoriske aminosyrereceptorantagonister eller midler, der forhindrer frie radikaler, være i stand til at forhindre eller forsinke sygdommens debut og progression. I laboratoriemodeller af amyotrofisk lateral sklerose er det blevet vist, at antioxidante stoffer og receptorantagonister (RAA'er) er i stand til at bremse sygdommens progression. Lignende tilgange kan være effektive ved Huntingtons sygdom. Kliniske forsøg med glutamatreceptorantagonister og stoffer, der forstærker funktionen af kompleks II i den mitokondrielle elektrontransportkæde, er i øjeblikket i gang.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]