Medicinsk ekspert af artiklen

Nye publikationer
Tricher Collins syndrom
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.

Når føtale lidelser knogle udviklingsprocesser har alvorlige kraniofacial deformation, og en af de sorter af sådan patologi er Treacher Collins syndrom (TCS) eller mandibulofastsialny, dvs. Maxillofacial dysostosis.
Kode for sygdommen ifølge ICD 10: klasse XVII (medfødte anomalier, deformationer og kromosomale abnormiteter), Q75.4 - mandibulofacial dysostose.
Årsager af Tricher Collins syndrom
Dette syndrom blev opkaldt fremragende britiske øjenlæge Edward Treacher Collins, som beskrev de vigtigste træk ved sygdommen mere end hundrede år siden. Men de europæiske læger ofte kalder denne form for anomalier facial knogler og kæbe sygdom eller syndrom Franceschetti - baseret på omfattende forskning af den schweiziske øjenlæge Adolf Franceschetti, der opfandt udtrykket "mandibulofastsialny dysostosis" i midten af sidste århundrede. Det medicinske samfund bruger også navnet - Franceschetti-Collins syndrom.
Årsager Treacher Collins syndrom - genmutationer TCOF1 (locus 5q31.3-33.3 kromosomer), der koder for et nucleolar phosphoprotein der er ansvarlig for dannelsen af kraniofaciale del af den menneskelige embryo. Som et resultat af et for tidlig fald i mængden af dette protein forstyrres biogenese og rRNA-funktioner. Ifølge genetikere Human Genome Research Program, disse processer føre til en reduktion i proliferationen af embryoniske neurale kamceller - valsen langs nerve tagrende, som i løbet af udvikling af embryoet i neuralrøret lukker.
Dannelse af den forreste del af kraniet væv opstår på grund af transformation og celledifferentiering øvre (hoved) del af crista neuralis, der migrerer langs neuralrøret til den første og anden Branchialfodder buer embryo. Og manglen på disse celler forårsager en kraniofaciale deformitet. Den kritiske periode for forekomst af misdannelser - fra 18 til 28 dage efter befrugtningen. Efter afslutning af migrationen af neurale kamceller (i den fjerde uge af svangerskabet) dannes næsten alle de løse mesenkymvæv i ansigtet, hvilket senere (5 til 8 uger) differentiere til skelet- og bindevæv i alle dele af ansigtet, halsen, hals, øre (herunder indenlandske) og fremtidige tænder.
Patogenese
Patogenesen af Treacher Collins syndrom er ofte køre i familier, og anomalien er nedarvet i en autosomal dominant princip, selv om der er tilfælde af autosomale recessive transmission defekter (mutationer ved andre gener, især POLR1C og POLR1D). Den mest uforudsigelige i maxillofacial dysostose er, at mutationen kun er arvet af børn i 40-48% af tilfældene. Det vil sige, 52-60% af patienterne forårsager Treacher Collins syndrom ikke forbundet med tilstedeværelsen af anomalier i familien og menes at have en patologi opstår fra sporadiske genetiske mutationer de novo. Mest sandsynligt repræsenterer de nye mutationer virkningerne af teratogene virkninger på fostret under graviditeten.
Blandt årsagerne til dette syndrom teratogene eksperter kalder høje doser af ethanol (ethylalkohol), stråling, cigaretrøg, tsitomegavirus og toxoplasmose, samt herbicider glyphosat (Raundalen, Glifor, Tornado, og andre.). Og listen over iatrogene faktorer var præparater fra seborrhea og akne med 13-cis-retinsyre (isotretinoin, Accutane); antikonvulsivt lægemiddel Phenytoin (Dilantin, Epanutin); psykotropiske stoffer Diazepam, Valium, Relan, Seduxen.
Symptomer af Tricher Collins syndrom
For det meste afhænger de kliniske tegn på mandibulofascial dysostose og graden af deres sværhedsgrad af funktionerne i manifestationen af genmutationer. Og de første tegn på denne anomali er i de fleste tilfælde synlige i barnet umiddelbart efter fødslen: Ansigtet med Tricher Collins syndrom har et karakteristisk udseende. Desuden er morfologiske anomalier normalt bilaterale og symmetriske.
De mest oplagte symptomer på Tricer Collins syndrom er:
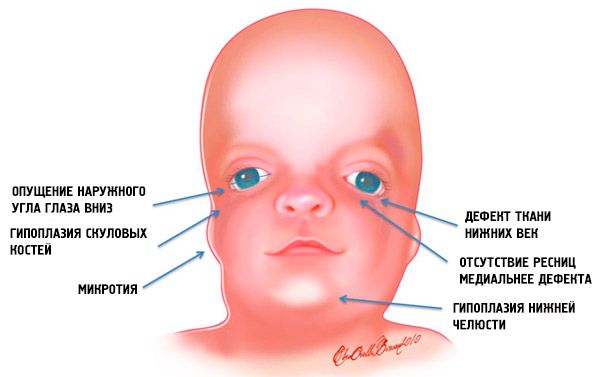
- underudvikling (hypoplasi) facial knoglerne i kraniet: zygomatic, zygomatic proces med forbenet, de laterale pterygoide plader, bihuler, mandibulære knogle epifyser og fremspring (kondylerne);
- underudvikling af knoglerne i underkæben (micrognathia) og mere stædig end normalt mandibulærvinklen;
- næse har en normal størrelse, men det ser ud til at være godt på grund af hypoplasia hos de superciliære buer og underudvikling eller mangel på zygomatiske buer i templernes område;
- øjets spalter er faldende, det vil sige at øjets snit er unormalt, med de ydre hjørner sænket nedad;
- defekter af nedre øjenlåg (coloboma) og delvis fravær af øjenvipper på dem;
- Øre med uregelmæssig form med en bred vifte af afvigelser, op til deres placering i hjørnet af underkæben, fraværet af lopper, blinde fistler mellem gedeøret og mundhjørnet mv.
- indsnævring eller infektion (atresi) af den ydre øregangskanal og anomalier i mellemøret
- fravær eller hypoplasi af parotid spytkirtler;
- pharyngeal hypoplasia (indsnævring af svælg og luftveje);
- ikke-helbredelse af den hårde gane (ulv mund), samt fraværet, forkortelse eller uigennemtrængelighed i den bløde gane.
Sådanne anatomiske abnormiteter har i alle tilfælde komplikationer. Disse er funktionelle hørselshemmede i form af ledende (ledende) høretab eller total døvhed; nedsat syn på grund af ukorrekt dannelse af øjne Galefejl forårsager problemer med fodring og indtagelse. Der er kæbrelaterede krænkelser af tændernes okklusion (en ukorrekt bid), hvilket igen forårsager problemer med tyggeri og artikulering. Patologier i den bløde gane forklarer næsestemmen.
Komplikationer og konsekvenser
Konsekvenserne af kraniofaciale anomalier i Treacher Collins syndrom manifesteret i det faktum, at ved fødslen hans normale intellektuelle evner, men på grund af at høre defekter og andre lidelser markeret sekundære mental retardering.
Derudover er børn med sådanne defekter akut opmærksomme på deres underlegenhed og lider, hvilket negativt påvirker deres nervesystem og psyke.
Diagnosticering af Tricher Collins syndrom
Postnatal diagnose af Tricher Collins syndrom er hovedsageligt baseret på kliniske tegn. Maxillofacial dysostose bestemmes let med syndromets fuldstændige ekspressivitet, men når der er minimal symptomer på patologi, kan der opstå problemer med formuleringen af den korrekte diagnose.
I dette tilfælde er der behov for særlig opmærksomhed for at vurdere alle de funktioner, der er forbundet med anomalier, især dem der påvirker vejrtrækningen (på grund af truslen om søvnapnø). Vurdering og overvågning af effektiviteten af fodring og mætning af hæmoglobin med oxygen udføres også.
I fremtiden - den 5.-6. Dag efter fødslen - er det nødvendigt at finde ud af graden af høreskader ved hjælp af audiologisk test, som skal udføres på barselshospitalet.
Der foretages en undersøgelse, hvor den instrumentelle diagnose udføres ved fluoroskopi af kranial-facial dysmorfologi; pantomografi (panorama røntgen af de benede strukturer i ansigtsskallen); fuld kraniel computertomografi i forskellige fremskrivninger; CT eller MR i hjernen for at bestemme tilstanden af den interne auditiv meatus.
Tidligste - Prænatal - diagnose af kraniofaciale anomalier i nærvær af Treacher Collins syndrom, familiens historie er mulig ved chorionvillus prøveudtagning på 10-11 uger af graviditeten (proceduren truer abort og infektion i livmoderen).
Også blodprøver af familiemedlemmer er taget; i 16-17 uger af graviditet er analysen af fostervæske (transabdominal amniocentese) taget; i 18-20 uger af graviditeten udføres fostoskopi, og blod tages fra placentas frugtbeholdere.
Men oftest i prænatal diagnose af dette syndrom bruger fosteret ultralyd (ved 20-24 ugers graviditet).
Hvilke tests er nødvendige?
Differential diagnose
Disse samme metoder eksperter bruge efter behov differentialdiagnose, at anerkende blødt udtalt Treacher Collins syndrom og skelne det fra andre medfødte abnormaliteter af kraniofaciale knogler, navnlig: Syndromes Apert, Crouzon, Nagera Peters-Hevelsa, Hellerman-Staefa samt med hemifaciale microsomia (Goldenhar syndrom), hypertelorism, tidlige uperforerede kranium suturer (craniostenosis) eller overtrædelse af fusion af ansigtets knogler (craniosynostosis).
Behandling af Tricher Collins syndrom
Som i alle tilfælde af genetisk betingede fødselsdefekter er behandling af Tricer Collins syndromet i svære former udelukkende palliativ, da der simpelthen ikke er nogen terapeutiske metoder til sådanne patologier. Spektret og graden af deformation i dette syndrom er omfattende, og derfor har arten og intensiteten af medicinsk intervention også en række muligheder.
Høreapparater bruges til at korrigere og forbedre hørelsen, for at forbedre taleklasserne med en taleterapeut.
Kirurgisk indgreb er påkrævet i en tidlig alder, i alvorlige tilfælde af luftveje indsnævring (trakeostomi udføres) og strubehovedet (udført gastrostommiya fodring) kan også kræve kirurgisk korrektion af ganen.
Operationer til forlængelse af underkæben udføres i en alder af 2-3 år eller derover. Rekonstruktion af blødt væv indbefatter korrektion af de nedre øjenlågskolonier og plastik i auriklerne.
Vejrudsigt
Hvad kan prognosen være for denne patologi? Det afhænger af graden af deformitet og intensiteten af symptomerne. Tricker Collins syndromet er en livslang diagnose.
 [25]
[25]