Medicinsk ekspert af artiklen

Nye publikationer
Subacut nekrotiserende encephalomyopati Leia
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
 [
[Årsager af Leias syndrom
Sygdommen er baseret på et enzym-mangel, giver uddannelse energi hovedsageligt ved stofskiftesygdomme pyrodruesyre og defekt elektron transport i respirationskæden. Pyruvatdehydrogenasekomplekset mangel udvikler (a-E1 underenhed), pyruvatcarboxylase, kompleks 1 (NAD coenzym Q-reduktase) og kompleks 4 (cytochromoxidase) respirationskæden.
Det konstateredes, at defekter pyruvat, komplekse 1 (NAD coenzym Q-reduktase) og kompleks 4 (cytochromoxidase) respirationskæden nedarves i en autosomal recessiv måde, defekter af pyruvatdehydrogenasekomplekset (a-E1 underenhed) - X-linked recessive. Da mtDNA punktmutationer, der påvirker 6-ATPase underenhed, mitokondrie arv karakteristisk. Oftest sker mistsens mutation forbundet med udskiftning af thymin til guanin eller cytosin i position 8993 mtDNA. Mindre almindelig er en mutation i position 9176 mtDNA. På grund af det faktum, at mutationer T8993G - grundlæggende defekt i syndromet NARP, beskrevet i familien med tilstedeværelsen af disse to sygdomme. Børn er også blevet beskrevet mtDNA-mutation i position 8344, som findes i syndromet MERRF.
Det foreslås, at der i tilfælde af akkumulering af mutant mtDNA i de fleste mitokondrier udvikles et alvorligt forløb af Leia syndromet. I den mitokondriegenese af denne tilstand detekteres mutant mtDNA i 90% af alle mitokondrier. Patogenese er forbundet med krænkelse af energiproduktion i celler og udvikling af mælkesyreoseose.
Symptomer af Leias syndrom
De første tegn på sygdommens debut i en tidlig alder (1-3 år). Men der er tilfælde af sygdomsmanifestationer i 2 uger og på 6-7 år. I første omgang udviklet uspecifikke lidelser: psykomotorisk retardering, tab af appetit, opkastning episoder, undervægtige. I de efterfølgende voksende neurologiske symptomer: muskulær hypotoni eller dystoni med overgangen hypertoni, kramper, myokloniske ryk eller tonisk-kloniske anfald, rysten af lemmer, choreoathetosis, koordinering lidelse, nedsat senereflekser, sløvhed, døsighed. Cerebral neurodegenerering har en progressiv karakter. Optagning symptomer på pyramideformede og ekstrapyramidale sygdom, nedsat synke. Ofte er der en sådan ændring af autoritet som ptose, oftalmoplegi, optisk atrofi, retinitis pigmentosa mindre. Nogle gange udvikler hypertrofisk kardiomyopati, der er episoder af takypnø.
Sjældent fortsætter sygdommen efter typen af akut encefalopati. Mere karakteristisk er en kronisk eller subakut strøm, hvilket fører til et fatalt udfald et par år efter sygdommens begyndelse. Med et hurtigt flow (adskillige uger) opstår døden som følge af lammelse af luftvejscentret.
Diagnosticering af Leias syndrom
I en biokemisk blodprøve påvises laktatacidose på grund af akkumulering af mælkesyre og pyrodruesyrer i blodet og væsken samt en forøgelse af indholdet af alanin i blodet. Også niveauet af ketonlegemer kan øges. I urinen er der øget udskillelse af organiske syrer: mælkesyre, fumarsyre osv. Karnitins niveau i blod og væv reduceres ofte.
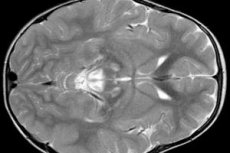
EEG-resultater afslører fokale tegn på epileptisk aktivitet. Ifølge MR-data påvises en udvidelse af hjernehvirvlerne, bilateral hjerneskade, forkalkning af de basale ganglier (caudatkernen, skalet, det sorte stof, den blegne bold). Det er også muligt at identificere atrofi af cerebrale halvkugler og hjerne stoffer.
Morfologiske undersøgelse viser grove ændringer i hjernens stof: symmetrisk nekrose, demyeliniserende og svampet degenerering af hjernen, primært midterste dele, bridge, basale ganglier, thalamus, synsnerven. Histologi involverer cystisk degeneration af hjernevæv, astrocytisk gliose, neuronal død, øge antallet af mitokondrier i cellerne. I skeletmuskler - ophobning af lipid indeslutninger falde histokemisk reaktion på komplekser af 1, 4 respiratoriske kæde af mitokondrier subsarkolemmalnoe overbelastning, unormale mitochondrier med afbrydelse af cristae. Fænomenet RRF registreres ofte ikke.
Hvordan man undersøger?
Hvilke tests er nødvendige?
Использованная литература