Medicinsk ekspert af artiklen

Nye publikationer
Apera syndrom
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.

 [
[Årsager aper's syndrom
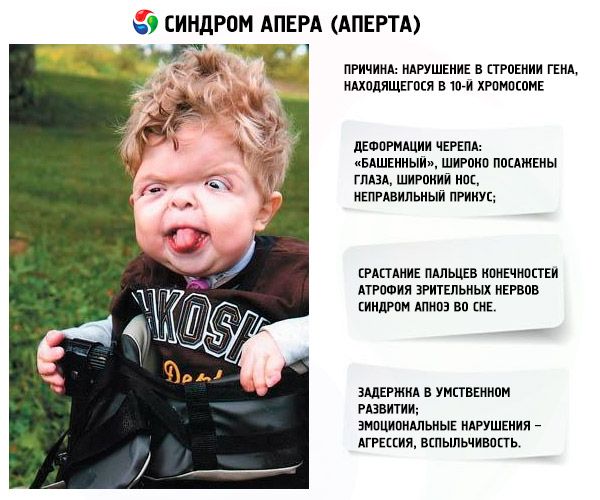
Udviklingen af syndromet fremkaldes af en overtrædelse i strukturen af genet, der ligger i det 10. Kromosom. Dette gen er ansvarlig for processen med adskillelse af fingrene og desuden for rettidig lukning af kraniale suturer.
Udover dette forårsager fremkomsten af patologien betragtede flyttet til drægtighedsperioden af smitsomme sygdomme (såsom tuberkulose eller rubella, syfilis eller influenza og meningitis), mor røntgenbestråling. Ofte findes et lignende syndrom hos børn født til ældre forældre.
Patogenese
Syndrom Apera overføres af arvelige stier. Hans type er autosomal dominant (dvs. I en familie, hvor en af forældrene er syg, er sandsynligheden for en baby med denne sygdom 50-100%).
Den unikke vækstfaktor for fibroblastreceptor 2 (FGFR2) på grund af mutation fører til en stigning i antallet af progenitorceller, som udvikles langs den osteogene vej. I sidste ende fører dette til øget dannelse af den subperiosteale knoglematrix og for tidlig senifikation af kraniale suturer under fosterudvikling. Sutursmeltningens rækkefølge og hastighed bestemmer graden af deformation og invaliditet. Efter at suturmaterialet vokser, bliver væksten af andre væv vinkelret på denne sutur begrænset, og de fusionerede knogler fungerer som et enkelt benskelet.
Den første genetiske beviser for, at syndactyly ved Apert syndrom er et resultat af keratinocytvækstfaktor-receptor defekt (KGFR) blev observation af en korrelation mellem ekspressionen af fibroblaster i KGFR og sværhedsgrad syndactyly.
Amblyopi og strabismus er mere almindelig hos patienter med mutationen FGFR2 Ser252Trp, og atrofi af optisk nerve disk er mere almindelig hos patienter med FGFR2 Pro253Arg mutationen. Hos patienter med FGR2 Ser252Trp har mutationer en signifikant højere forekomst af synshæmning sammenlignet med patienter med FGFR2 Pro253Arg mutation.
Symptomer aper's syndrom
Individuelle manifestationer af sygdommen er tydeligt synlige selv ved barnets fødsel, fordi de udvikler sig selv indenfor moderens livmoder. Blandt de vigtigste symptomer på syndromet:
- Kraniet er deformeret - strækkes til højden og bliver en slags "tårn"; Desuden er øjnene bredt plantet og lidt fremspringende (fordi størrelsen på øjenbanen falder); næsen udvides og en ukorrekt bid formes (de øvre tænder rager ud);
- Tæerne på lemmerne er fuldt fusionerede (for det meste umærkede og også midt i indekset) og ligner en hudmembran eller fuld fusion af knogler; Derudover kan ekstra fingre vokse;
- Forsinkelse i mental udvikling (slet ikke);
- De optiske nerver er atrofierede, hvilket medfører, at synsstyrken falder (i nogle tilfælde udvikles en fuldstændig mangel på syn);
- Forøget intrakranielt tryk som følge af for tidlig overvævning af kraniale suturer - manifesterer som hovedpine og opkastning med kvalme;
- Da overkæben har været underudviklet, er der problemer med vejrtrækning;
- Søvnapnø syndrom er almindeligt.
- Emosionelle manifestationer - aggression, manglende tilbageholdenhed, stærkt humør.
Komplikationer og konsekvenser
Diagnosticering aper's syndrom
Følgende foranstaltninger er nødvendige for diagnosen:
- Lægen skal analysere patientens klager samt medicinsk historie. Det er nødvendigt at finde ud af, om der er tilfælde af udvikling af en sådan patologi i familien;
- Neurologisk undersøgelse for at vurdere formlen på kraniet og patientens intellektuelle udvikling (specielle spørgeskemaer samt samtale);
- Undersøgelse af fundus for at identificere forekomsten af symptomer på øget intrakranielt tryk (ødem DZN samt blørhed i kanterne);
- For at vurdere tilstanden af kraniet udføres dets radiografi;
- Computeren, samt magnetisk resonans imaging hoved til lagvis undersøge den cerebrale struktur af kraniet, for at bestemme tilstedeværelsen af symptomer på tidlig sammensmeltning af de kranielle suturer, men derudover hydrocephalus (på grund af forøget intrakranielt tryk akkumulerer overskydende væske (dette cerebrospinalvæske, der fremmer udvekslingen proces stoffer samt ernæring af hjernen));
- Røntgen af fødderne med børster for at finde årsagen, på grund af hvilken der var en fusion af fingre (dette er vigtigt for planlægning efterfølgende kirurgisk indgreb);
- En rådgivning med en medicinsk genetiker og en neurokirurg kan foreskrives.
Analyser
Genetisk analyse af hyppige mutationer forekommende i genet af typen FGFR2 udføres.
Differential diagnose
Differentier dette syndrom med andre genetiske patologier, hvor craniosynostose observeres. Disse er sygdomme som Pfeiffer, Cruson og Sethra-Chotzen og Carpenter syndromer. For at udelukke disse anomalier anvendes molekylære genetiske testmetoder.
Hvem skal kontakte?
Behandling aper's syndrom
Udførelse af kirurgisk operation anses for at være den eneste effektive metode til behandling af Apera syndromet - det hjælper med at rette individuelle fysiske defekter og korrigerer også tilbagegangen i mental udvikling.
Under processen med denne procedure udføres lukkelse af koronal sutur for at forhindre mulig trauma til hjernen. Den mest almindelige metode er craniofacial distraktion, hvor graden af kraniet udføres. For at fjerne individuelle defekter i ansigtet udføres en ortodontisk og / eller ortognatisk operation.
Desuden fjerner patienterne kirurgisk fusion af fingrene.