Medicinsk ekspert af artiklen

Nye publikationer
T-celle-lymfomer i huden
Last reviewed: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
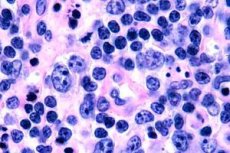
T-celle lymfomer registreres oftest hos ældre mennesker, selvom isolerede tilfælde af sygdommen også ses hos børn. Mænd er syge dobbelt så ofte som kvinder. T-celle lymfomer er epidermotrope af natur.
Årsager T-celle-lymfomer i huden
Årsagerne til og patogenesen af kutane T-celle lymfomer er ikke fuldt ud forstået. I øjeblikket anser de fleste forskere human T-celle leukæmivirus type 1 (HTLV-1) I for at være den primære ætiologiske faktor, der initierer udviklingen af maligne T-celle lymfomer i huden. Samtidig diskuteres rollen af andre vira i udviklingen af T-celle lymfom: Epstein-Barr-virus, herpes simplex type 6. Hos patienter med T-celle lymfom findes vira i huden, perifert blod og Langerhans-celler. Antistoffer mod HTLV-I påvises hos mange patienter med mycosis fungoides.
En vigtig plads i patogenesen af T-cellelymfomer spilles af immunopatologiske processer i huden, hvoraf den vigtigste er den ukontrollerede proliferation af klonale lymfocytter.
Cytokiner produceret af lymfocytter, epitelceller og celler i makrofagsystemet har proinflammatoriske og proliferative virkninger (IL-1, ansvarlig for lymfocytdifferentiering; IL-2 - T-cellevækstfaktor; IL-4 og IL-5, der øger tilstrømningen af eosinofiler i læsionen og deres aktivering osv.). Som følge af tilstrømningen af T-lymfocytter i læsionen dannes Pautrier-mikroabscesser. Samtidig med stigningen i lymfocytproliferation undertrykkes aktiviteten af antitumorforsvarsceller: naturlige dræbere, lymfocyttoksiske lymfocytter, dendritiske celler, især Langerhanske celler, samt cytokiner (IL-7, IL-15 osv.) - tumorvæksthæmmere. Arvelige faktorers rolle kan ikke udelukkes. Tilstedeværelsen af familiære tilfælde og hyppig påvisning af visse histokompatibilitetsantigener (HLA B-5 og HLA B-35 - ved højmaligne hudlymfomer, HLA A-10 - ved mindre aggressive lymfomer, HLA B-8 - ved den erythrodermiske form af mycosis fungoides) bekræfter dermatosens arvelige natur.
Kliniske observationer indikerer en mulig transformation af langvarige kroniske dermatoser (neurodermatitis, atopisk dermatitis, psoriasis osv.) til mycosis fungoides. Nøglefaktoren er lymfocytternes langvarige persistens i inflammationsfokus, hvilket forstyrrer immunovervågningen og fremmer fremkomsten af en klon af maligne lymfocytter og dermed udviklingen af en malign proliferativ proces.
Påvirkningen af fysiske faktorer på kroppen, såsom solindstråling, ioniserende stråling og kemiske stoffer, kan føre til fremkomsten af en klon af "genotraumatiske" lymfocytter, der har en mutagen effekt på lymfoide celler og udvikling af lymfocytmalignitet.
Derfor kan T-cellelymfomer betragtes som en multifaktoriel sygdom, der begynder med aktivering af lymfocytter under påvirkning af forskellige kræftfremkaldende, "genotraumatiserende" faktorer og fremkomsten af en dominant T-celleklon. Sværhedsgraden af immunovervågningsforstyrrelsen, klonen af maligne lymfocytter, bestemmer de kliniske manifestationer (plettede, plak- eller tumorelementer) af T-cellelymfomer.
Patogenese
I det tidlige stadie af mycosis fungoides observeres akantose med brede udløbere, hyperplasi og kompaktering af basale keratinocytter, vakuolær degeneration af nogle basalceller, atypiske mitoser i forskellige lag af epidermis, epidermotropisme af infiltratet med penetration af lymfocytter ind i epidermis. I dermis observeres små infiltrater omkring karrene, bestående af enkelte mononukleære celler med hyperkrome kerner - "mykotiske" celler. I det andet stadie observeres en stigning i sværhedsgraden af det dermale infiltrat og epidermotropisme af infiltratcellerne, hvorved maligne lymfocytter trænger ind i epidermis og danner klynger i form af Potrier-mikroabscesser. I det tredje, tumorstadiet, observeres massiv akantose og mindre atrofi af epidermis, samt øget infiltration af epidermis af tumorlymfocytter, som danner flere Potrier-mikroabscesser. Det massive infiltrat er placeret i hele dermis' tykkelse og dækker en del af hypodermis. Der observeres blastformer af lymfocytter.
Kutant stort anaplastisk T-celle lymfom
Det er repræsenteret af en gruppe lymfoproliferative processer, der er karakteriseret ved tilstedeværelsen af proliferater fra atypiske klonale store anaplastiske CD30+ T-celler. Som regel udvikler det sig sekundært i tumorstadiet af mycosis fungoides eller i Sezary syndrom, men kan udvikle sig uafhængigt eller med spredning af systemiske lymfomer af denne type. Klinisk svarer sådanne lymfomer til den såkaldte dekapiterede form af mycosis fungoides i form af enkelte eller flere lymfomer, normalt grupperede.
Histologisk optager proliferatet næsten hele dermis med eller uden epidermotropisme i tilfælde af epidermal atrofi.
Cytologisk kan tumorceller variere i størrelse og form. Baseret på disse egenskaber skelnes der mellem mellem- og storcellet pleomorft T-cellelymfom med kerner af forskellige uregelmæssige konfigurationer - snoede, multilobede, med tæt kromatin, en veldefineret nukleol og forholdsvis rigeligt cytoplasma; immunoblastisk - med store runde eller ovale kerner med tydelig karyoplasma og én centralt placeret nukleol; anaplastisk - med grimme, meget store celler med kerner af uregelmæssig konfiguration og rigeligt cytoplasma. Fænotypisk tilhører hele denne gruppe T-hjælperlymfomer og kan være CD30+ eller CD30-.
R. Willemze et al. (1994) viste, at forløbet af CD30+ lymfom er mere gunstigt. Genotypisk detekteres klonal rearrangement af T-lymfocytreceptoren.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Symptomer T-celle-lymfomer i huden
Den mest almindelige sygdom i gruppen af T-celle lymfomer i huden er mycosis fungoides, som tegner sig for omkring 70% af tilfældene. Der er tre kliniske former for sygdommen: klassisk, erythrodermisk og halshugget. T-celle lymfomer er karakteriseret ved polymorfi af udslæt i form af pletter, plaques, tumorer.
Den erytrodermiske form af mycosis fungoides begynder normalt med ukontrollerbar kløe, hævelse, universel hyperæmi, forekomsten af erytematøse-pladeepitellæsioner på huden på kroppen og ekstremiteterne, som har tendens til at smelte sammen og udvikle erytrodermi inden for 1-2 måneder. Næsten alle patienter har palmar-plantar hyperkeratose og diffus udtynding af hår over hele huden. Alle grupper af lymfeknuder er stærkt forstørrede. Forstørrede inguinale, femorale, aksillære, cubitale lymfeknuder palperes som "pakker" af tæt elastisk konsistens, ikke fusioneret med det omgivende væv, smertefrit. Den generelle tilstand forværres kraftigt: feber med en kropstemperatur på op til 38-39 ° C, nattesved, svaghed og vægttab forekommer. I øjeblikket betragtes Sezary syndrom af mange dermatologer som den sjældneste leukæmiske variant af den erytrodermiske form af mycosis fungoides,
En udtalt leukocytose ses i lymfocytogrammer - Sezary-celler. Sezary-celler er maligne T-hjælpere, hvis kerner har en foldet cerebriform overflade med dybe invaginationer af kernemembranen. Et dødeligt udfald ses efter 2-5 år, hvis hyppige årsag er kardiovaskulær patologi og forgiftning.
Den halshuggede form af mycosis fungoides er karakteriseret ved den hurtige udvikling af tumorlignende læsioner på tilsyneladende sund hud uden forudgående langvarig plakdannelse. Denne form er karakteriseret ved en høj grad af malignitet, hvilket betragtes som en manifestation af lymfosarkom. Et dødeligt udfald observeres inden for et år.
Niveauer
Den klassiske form for mycosis fungoides er karakteriseret ved tre udviklingsstadier: erythematøs-pladeepitel, plak og tumor.
Det første stadie ligner det kliniske billede af nogle godartede inflammatoriske dermatoser - eksem, seboroisk dermatitis, plaque parapsoriasis. På dette stadie af sygdommen observeres pletter i forskellige størrelser, intenst lyserøde, lyserød-røde med et lilla skær, runde eller ovale konturer, med relativt klare grænser, overfladisk klidlignende eller finpladeformet afskalning. Elementerne er ofte placeret på forskellige områder af huden, oftest på kroppen og ansigtet. Gradvist stiger deres antal. Over tid kan processen antage karakter af erythrodermi (erytrodermisk stadie). Udslættet kan eksistere i årevis eller forsvinde spontant. I modsætning til godartede inflammatoriske dermatoser er elementerne af udslæt og kløe på dette stadie resistente over for behandlingen.
Infiltrativ plakfase udvikler sig over flere år. I stedet for tidligere eksisterende plettede udslæt fremkommer der plaques med runde eller uregelmæssige konturer, intenst lilla i farven, tydeligt afgrænset fra sund hud, tætte og med en skællende overflade. Deres konsistens ligner "tyk pap". Nogle af dem forsvinder spontant og efterlader områder med mørkebrun hyperpigmentering og/eller atrofi (poikiloderma). Kløe på dette stadie er endnu mere intens og smertefuld, feber og vægttab observeres. Lymfadenopati kan observeres på dette stadie.
I det tredje, tumorstadiet, opstår smertefri tumorer med en tæt, elastisk konsistens, gulrød farve, der udvikler sig fra plaques eller opstår på tilsyneladende sund hud. Tumorernes form er sfærisk eller fladtrykt og ligner ofte en svampehat. Tumorer kan opstå overalt. Deres antal varierer meget fra enkeltstående til dusinvis, størrelser - fra 1 til 20 cm i diameter. Når længe eksisterende tumorer opløses, dannes der sår med ujævne kanter og en dyb bund, der når fascia eller knoglen. Lymfeknuder, milt, lever og lunger påvirkes oftest. Den generelle tilstand forværres, der opstår og øges symptomer på forgiftning, og svaghed udvikles. Den gennemsnitlige levetid for patienter med den klassiske form for mycosis fungoides fra diagnosetidspunktet er fra 5 til 10 år. Dødelighed observeres normalt fra interkurrente sygdomme: lungebetændelse, hjerte-kar-svigt, amyloidose. Subjektivt mærkes kløe, og når tumorer opløses, smerter i de berørte områder.
Hvad skal man undersøge?
Hvordan man undersøger?
Behandling T-celle-lymfomer i huden
I det erytematøse-pladecellestadium behøver patienter ikke antitumorbehandling; de får ordineret topiske kortikosteroider (prednisolon, betamethason, dexamethasonderivater), interferon alfa (3 millioner IE dagligt, derefter 3 gange om ugen i 3-6 måneder afhængigt af kliniske manifestationer eller behandlingseffektivitet), interferon gamma (100.000 IE dagligt i 10 dage, cyklussen gentages 12-3 gange med en 10-dages pause), PUVA-terapi eller Re-PUVA-terapi. Effektiviteten af PUVA-terapi er baseret på den selektive dannelse af kovalente tværbindinger af psoralener med DNA i prolifererende T-hjælperceller, hvilket hæmmer deres deling. I andet trin anvendes der ud over de ovennævnte midler systemiske kortikosteroider (30-40 mg prednisolon dagligt i 1,5-2 måneder) og cytostatika (prospedin 100 mg dagligt, i alt 4-5 injektioner). Kombination af interferoner med andre behandlingsmetoder har en mere udtalt terapeutisk effekt (interferoner + PUVA, interferoner + cytostatika, interferoner + aromatiske retinoider).
I tumorstadiet er den primære metode polykemoterapi. Der anvendes en kombination af vincristin (0,5-1 mg intravenøst én gang dagligt, i alt 4-5 injektioner) med prednisolon (40-60 mg oralt dagligt under kemoterapi), prospidin (100 mg dagligt, i alt 3 g) og interferoner. Fotodynamisk, elektronstråleterapi og fotoferese (ekstrakorporal fotokemoterapi) anbefales.