Medicinsk ekspert af artiklen

Nye publikationer
Keratodermi: årsager, symptomer, diagnose, behandling
Sidst revideret: 07.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
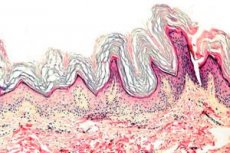
Keratodermi er en gruppe af dermatoser, der er karakteriseret ved en forstyrrelse af keratiniseringsprocessen - overdreven horndannelse hovedsageligt på håndflader og fodsåler.
Årsagerne til og patogenesen af sygdommen er ikke fuldt ud klarlagt. Forskning har fastslået, at keratodermier skyldes mutationer i generne, der koder for keratin 6, 9, 16. A-vitaminmangel, hormonelle dysfunktioner, primært i kønskirtlerne, samt bakterielle og virale infektioner er af stor betydning i patogenesen. De er et af symptomerne på arvelige sygdomme og tumorer i indre organer (parapsoriatiske keratodermier).
Symptomer. Der skelnes mellem diffus (Unna-Tost keratodermi, Meleda keratodermi, Papillon-Lefèvre keratodermi, mutilerende keratodermi og syndromer, der inkluderer diffus keratodermi som et af hovedsymptomerne) og fokal (dissemineret plettet keratodermi af Fischer-Buschke, akrokeratoelastoidose af Kosti, begrænset keratodermi af Bruhauer-Franzesthesti, lineær keratodermi af Fuchs osv.) keratodermi.
Winy-Tost keratodermi (synonymer: medfødt iktyose i håndflader og fodsåler, Winy-Tost syndrom) overføres autosomalt dominant. Der er en diffus overdreven keratinisering af huden på håndflader og fodsåler (nogle gange kun fodsåler), som udvikler sig i de første to leveår. Den patologiske hudproces begynder med en let fortykkelse af huden på håndflader og fodsåler i form af en strimmel af erytem med en lys farve på grænsen til sund hud. Med tiden opstår glatte, gullige, hornede lag på deres overflade. Læsionen spreder sig sjældent til håndleddenes eller fingrenes ryg. Hos nogle patienter kan der dannes overfladiske eller dybe revner, og lokal hyperhidrose observeres. Hos den patient, som forfatteren observerede, led onkelen på mors side, broren og sønnen af Winy-Tost keratodermi.
Der beskrives tilfælde af skader på negle (fortykkelse), tænder og hår ved Winy-Tost keratodermi i kombination med forskellige skeletanomalier og patologier i indre organer, nerve- og endokrine systemer.
Histopatologi. Histologisk undersøgelse afslører markant hyperkeratose, granulose, akantose og små inflammatoriske infiltrater i den øvre dermis. Differentialdiagnose. Sygdommen skal differentieres fra andre typer keratodermi.
Meleda keratodermi (synonymer: Meleda sygdom, medfødt progressivt akrokeratom, Siemens' palmoplantar transgradient keratose, Kogoys arvelige palmoplantar progressive keratose) nedarves autosomal recessivt. Denne form for keratodermi er karakteriseret ved tykke, gulbrune hornlag med dybe revner. En violet-lilla kant på flere millimeters bredde er synlig langs læsionens kanter. Processen spreder sig typisk til bagsiden af hænder og fødder, underarme og skinneben. De fleste patienter oplever lokal hyperhidrose. I denne forbindelse bliver overfladen af håndflader og fodsåler let fugtig og dækket af sorte prikker (svedkirtelkanaler).
Sygdommen kan udvikle sig i 15-20-årsalderen. Neglene bliver tykkere og deformeret.
Histopatologi. Histologisk undersøgelse afslører hyperkeratose, undertiden akantose, og et kronisk inflammatorisk infiltrat i den papillære dermis.
Differentialdiagnose. Melela keratodermi skal skelnes fra Unna-Tost keratodermi.
Keratoderma Papillon-Lefevre (synonym: palmoplantar hyperkeratose med parodontitis) nedarves autosomal recessivt.
Sygdommen manifesterer sig i 2.-3. leveår. Det kliniske billede af sygdommen ligner Melelas sygdom. Derudover er der karakteristiske ændringer i tænderne (unormaliteter i fremkomsten af mælketænder og blivende tænder med udvikling af karies, gingivitis, hurtigt fremadskridende parodontose med for tidligt tandtab).
Histopatologi. Histologisk undersøgelse afslører fortykkelse af alle lag af epidermis, især hornlaget, og ubetydelige cellulære klynger af lymfocytter og histiocytter i dermis.
Differentialdiagnose. Sygdommen bør skelnes fra andre keratodermier. Et vigtigt kendetegn er den karakteristiske tandpatologi, som ikke findes ved andre former for arvelig diffus keratodermi.
Keratoderma mutilans (synonymer: Fonwinkel syndrom, arveligt mutilerende keratom) er en type diffus keratodermi, der nedarves autosomalt dominant. Den udvikler sig i det 2. leveår og er karakteriseret ved diffuse hornaflejringer på huden i håndflader og fodsåler med hyperhidrose. Med tiden dannes der snorlignende riller på fingrene, hvilket fører til kontrakturer og spontan amputation af fingrene. Follikulær keratose udtrykkes på håndryggen samt i området omkring albue- og knæleddene. Neglepladerne ændres (ofte som urglas). Tilfælde af hypogonadisme, rubinrød alopeci, høretab og pachyonyki er blevet beskrevet.
Histopatologi. Histologisk undersøgelse afslører svær hyperkeratose, granulose, akantose og små inflammatoriske infiltrater i dermis, bestående af lymfocytter og histiocytter.
Differentialdiagnose. Når man skelner mellem mutilerende keratodermi og andre former for diffus keratodermi, bør man først og fremmest tage hensyn til mutilationseffekten, som ikke er typisk for andre former. Ved differentialdiagnostik af alle former for diffus keratodermi er det nødvendigt at huske, at det kan være et af hovedsymptomerne på en række arvelige syndromer.
Behandling. Neotigazon er indiceret i den generelle behandling af keratodermi. Dosis af lægemidlet afhænger af processens sværhedsgrad og er 0,3-1 mg/kg af patientens vægt. I fravær af neotigazon anbefales vitamin A i en dosis på 100 til 300.000 mg pr. dag i lang tid. Ekstern terapi består af brug af salver med aromatiske retinoider, keratolytiske og steroide midler.
 [
[ Hvad generer dig?
Hvad skal man undersøge?
Hvordan man undersøger?