Medicinsk ekspert af artiklen

Nye publikationer
Arvelig nefritis (Alports syndrom) hos børn
Sidst revideret: 05.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
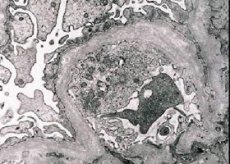
Arvelig nefritis (Alports syndrom) er en genetisk bestemt arvelig ikke-immun glomerulopati, der manifesterer sig ved hæmaturi (undertiden med proteinuri), progressiv nedgang i nyrefunktionen med udvikling af kronisk nyresvigt, ofte kombineret med sensorineural døvhed og synshandicap.
Sygdommen blev først beskrevet i 1902 af LG Guthrie, som observerede en familie, hvor hæmaturi blev observeret i flere generationer. I 1915 beskrev AF Hurst udviklingen af uræmi hos medlemmer af samme familie. I 1927 identificerede A. Alport først høretab hos flere slægtninge med hæmaturi. I 1950'erne blev øjenlæsioner ved en lignende sygdom beskrevet. I 1972 afslørede Hinglais et al. under en morfologisk undersøgelse af nyrevæv ujævn ekspansion og lagdeling af de glomerulære basalmembraner hos patienter med arvelig hæmaturi. I 1985 blev det genetiske grundlag for arvelig nefritis identificeret - en mutation i type IV-kollagengenet (Fiengold et al., 1985).
Undersøgelsen af sygdommens genetiske natur tillod os at konkludere, at forskellene i de fænotypiske manifestationer af arvelig nefritis (med eller uden høretab) skyldes graden af ekspression af det mutante gen. Således betragtes alle kliniske varianter i øjeblikket som manifestationer af én sygdom, og udtrykket "arvelig nefritis" er synonymt med udtrykket "Alport syndrom".
Ifølge epidemiologiske undersøgelser forekommer arvelig nefritis med en hyppighed på 17 pr. 100.000 børn.
 [
[ Årsager til Alports syndrom
Det genetiske grundlag for sygdommen er en mutation i genet for a-5-kæden af type IV-kollagen. Denne type er universel for basalmembranerne i nyrerne, cochlearapparatet, linsekapslen, nethinden og hornhinden i øjet, hvilket er blevet bevist i studier med monoklonale antistoffer mod denne kollagenfraktion. For nylig er muligheden for at bruge DNA-prober til prænatal diagnostik af arvelig nefritis blevet indikeret.
Det understreges, at det er vigtigt at teste alle familiemedlemmer med DNA-sonder for at identificere bærere af det mutante gen, hvilket er af stor betydning i forbindelse med medicinsk og genetisk rådgivning af familier med denne sygdom. Op til 20 % af familierne har dog ingen slægtninge, der lider af nyresygdom, hvilket tyder på en høj frekvens af spontane mutationer af det unormale gen. De fleste patienter med arvelig nefritis har personer med nyresygdom, høretab og synspatologi i deres familier; blodsbeslægtede ægteskaber mellem personer med en eller flere forfædre er vigtige, da sandsynligheden for at modtage de samme gener fra begge forældre øges i ægteskaber mellem beslægtede individer. Autosomalt dominante, autosomalt recessive og dominante, X-bundne transmissionsveje er blevet etableret.
Hos børn skelnes der oftest mellem tre typer arvelig nefritis: Alports syndrom, arvelig nefritis uden høretab og familiær benign hæmaturi.
Alports syndrom er en arvelig nefritis med hørenedsættelse. Den er baseret på en kombineret defekt i strukturen af kollagenet i den glomerulære basalmembran i nyrerne, øret og øjet. Genet for klassisk Alports syndrom er placeret i locus 21-22q på den lange arm af X-kromosomet. I de fleste tilfælde nedarves det dominant, knyttet til X-kromosomet. I denne henseende er Alports syndrom mere alvorligt hos mænd, da funktionen af det mutante gen hos kvinder kompenseres af en sund allel af det andet, ubeskadigede kromosom.
Det genetiske grundlag for udviklingen af arvelig nefritis er mutationer i generne for alfakæderne i type IV-kollagen. Seks alfakæder af type IV-kollagen G er kendte: generne for a5- og a6-kæderne (Col4A5 og Col4A5) er placeret på den lange arm af X-kromosomet i 21-22q-zonen; generne for a3- og a4-kæderne (Col4A3 og Col4A4) er på det 2. kromosom; generne for a1- og a2-kæderne (Col4A1 og Col4A2) er på det 13. kromosom.
I de fleste tilfælde (80-85%) påvises et X-bundet arvemønster af sygdommen, forbundet med skade på Col4A5-genet som følge af deletion, punktmutationer eller splejsningsforstyrrelser. I øjeblikket er der fundet mere end 200 mutationer af Col4A5-genet, som er ansvarlige for forstyrrelsen af syntesen af a5-kæderne i type IV-kollagen. Ved denne type arv manifesterer sygdommen sig hos børn af begge køn, men hos drenge er den mere alvorlig.
Mutationer i loci for Col4A3- og Col4A4-generne, der er ansvarlige for syntesen af a3- og a4-kæderne af type IV-kollagen, nedarves autosomalt. Ifølge forskning observeres den autosomalt dominante type arv i 16% af tilfældene af arvelig nefritis, og den autosomalt recessive type observeres hos 6% af patienterne. Omkring 10 varianter af mutationer af Col4A3- og Col4A4-generne er kendte.
Resultatet af mutationer er en forstyrrelse af samlingsprocesserne for type IV-kollagen, hvilket fører til en forstyrrelse af dets struktur. Type IV-kollagen er en af hovedkomponenterne i den glomerulære basalmembran, cochlearapparatet og øjets linse, hvis patologi vil blive påvist i klinikken for arvelig nefritis.
Kollagen type IV, som er en del af den glomerulære basalmembran, består hovedsageligt af to a1-kæder (IV) og én a2-kæde (IV) og indeholder også a3-, a4- og a5-kæder. Oftest, ved X-bundet arv, ledsages mutationen af Col4A5-genet af fraværet af a3-, a4-, a5- og a6-kæder i strukturen af kollagen type IV, og antallet af o1- og a2-kæder i den glomerulære basalmembran stiger. Mekanismen bag dette fænomen er uklar, det antages, at årsagen er posttranskriptionelle ændringer i mRNA.
Fraværet af a3-, a4- og a5-kæder i strukturen af type IV-kollagen i de glomerulære basalmembraner fører til deres udtynding og skrøbelighed i de tidlige stadier af Alports syndrom, som klinisk manifesterer sig oftere ved hæmaturi (sjældnere ved hæmaturi med proteinuri eller kun proteinuri), høretab og lenticonus. Yderligere progression af sygdommen fører til fortykkelse og nedsat permeabilitet af basalmembranerne i de sene stadier af sygdommen, med proliferation af kollagen af type V og VI i dem, manifesteret i en stigning i proteinuri og et fald i nyrefunktionen.
Arten af den mutation, der ligger til grund for arvelig nefritis, bestemmer i høj grad dens fænotypiske manifestation. I tilfælde af deletion af X-kromosomet med samtidig mutation af Col4A5- og Col4A6-generne, der er ansvarlige for syntesen af a5- og a6-kæderne af type IV-kollagen, er Alports syndrom kombineret med leiomyomatose i spiserøret og kønsorganerne. Ifølge forskningsdata ses en større sværhedsgrad af den patologiske proces i tilfælde af en mutation af Col4A5-genet forbundet med en deletion, en kombination af nyreskade med ekstrarenale manifestationer og tidlig udvikling af kronisk nyresvigt sammenlignet med en punktmutation af dette gen.
Morfologisk afslører elektronmikroskopi udtynding og lagdeling af de glomerulære basalmembraner (især lamina densa) og tilstedeværelsen af elektrontætte granuler. Glomerulære læsioner kan være heterogene hos den samme patient, fra minimale fokale mesangiale læsioner til glomerulosklerose. Glomerulitis ved Alport syndrom er altid immunonegativ, hvilket adskiller det fra glomerulonefritis. Karakteristiske træk omfatter udvikling af tubulær atrofi, lymfohistiocytisk infiltration og tilstedeværelsen af "skumceller" med lipidindeslutninger - lipofager. Efterhånden som sygdommen skrider frem, afsløres fortykkelse og udtalt ødelæggelse af de glomerulære basalmembraner.
Visse ændringer i immunsystemet afsløres. Patienter med arvelig nefritis har et nedsat IgA-niveau og en tendens til at øge IgM-koncentrationen i blodet. IgG-niveauet kan være forhøjet i de tidlige stadier af sygdommen og falde i de senere stadier. Måske er stigningen i IgM- og G-koncentrationen en slags kompenserende reaktion som reaktion på IgA-mangel.
Den funktionelle aktivitet af T-lymfocytsystemet reduceres; et selektivt fald i B-lymfocytter, der er ansvarlige for syntesen af Ig A, observeres, den fagocytiske forbindelse af immunitet forstyrres, hovedsageligt på grund af forstyrrelse af kemotaksi og intracellulære fordøjelsesprocesser i neutrofiler.
Ved undersøgelse af en nyrebiopsi hos patienter med Alports syndrom afslører elektronmikroskopidata ultrastrukturelle ændringer i den glomerulære basalmembran: udtynding, strukturforstyrrelse og opsplitning af de glomerulære basalmembraner med en ændring i dens tykkelse og ujævne konturer. I de tidlige stadier af arvelig nefritis bestemmer defekten udtyndingen og skrøbeligheden af de glomerulære basalmembraner.
Udtynding af glomerulære membraner er et mere gunstigt tegn og er mere almindeligt hos piger. Et mere konstant elektronmikroskopisk tegn ved arvelig nefritis er opsplitning af basalmembranen, og sværhedsgraden af dens ødelæggelse korrelerer med processens sværhedsgrad.
Symptomer på Alports syndrom hos børn
De første symptomer på Alports syndrom i form af isoleret urinsyndrom opdages oftest hos børn i de første tre leveår. I de fleste tilfælde opdages sygdommen tilfældigt. Urinsyndrom opdages under en forebyggende undersøgelse af barnet, før indlæggelse på en børnepasningsinstitution eller under ARVI. I tilfælde af patologi i urinen under ARVI. Ved arvelig nefritis er der, i modsætning til erhvervet glomerulonefritis, ingen latent periode.
I sygdommens indledende fase lider barnets helbred kun lidt, et karakteristisk træk er urinsyndromets vedvarende og modstandsdygtighed. Et af hovedtegnene er hæmaturi af varierende sværhedsgrad, observeret i 100% af tilfældene. En stigning i graden af hæmaturi ses under eller efter luftvejsinfektioner, fysisk aktivitet eller efter forebyggende vaccinationer. Proteinuri overstiger i de fleste tilfælde ikke 1 g/dag, i begyndelsen af sygdommen kan være ustabil, og efterhånden som processen skrider frem, stiger proteinuri. Med jævne mellemrum kan leukocyturi med en overvægt af lymfocytter være til stede i urinsedimentet, hvilket er forbundet med udviklingen af interstitielle forandringer.
Efterfølgende er delvis nyrefunktion nedsat, patientens generelle tilstand forværres: forgiftning, muskelsvaghed, arteriel hypotension, ofte hørenedsættelse (især hos drenge) og undertiden synshandicap optræder. Forgiftning manifesterer sig ved bleghed, træthed og hovedpine. I sygdommens indledende fase opdages høretab i de fleste tilfælde kun ved audiografi. Høretab ved Alports syndrom kan forekomme i forskellige perioder af barndommen, men oftest diagnosticeres høretab i alderen 6-10 år. Høretab hos børn begynder med høje frekvenser og når en betydelig grad i luft- og benledning, og går fra lydledende til lydopfattende høretab. Høretab kan være et af de første symptomer på sygdommen og kan gå forud for urinvejssyndrom.
I 20% af tilfældene oplever patienter med Alport syndrom ændringer i synsorganerne. De hyppigst opdagede anomalier er linsens: sfærofoki, anterior, posterior eller blandet lenticonus og forskellige grå stær. I familier med Alport syndrom er der en betydelig hyppighed af myopi. En række forskere bemærker konstant bilaterale perimakulære forandringer i disse familier i form af lyse hvidlige eller gullige granuleringer i corpus luteum. De anser dette tegn for at være et konstant symptom, der har høj diagnostisk værdi ved Alport syndrom. KS Chugh et al. (1993) fandt i en oftalmologisk undersøgelse hos patienter med Alport syndrom et fald i synsstyrken i 66,7% af tilfældene, anterior lenticonus i 37,8%, retinale pletter i 22,2%, grå stær i 20% og keratokonus i 6,7%.
Hos nogle børn med arvelig nefritis, især når nyresvigt udvikler sig, ses en betydelig forsinkelse i den fysiske udvikling. Efterhånden som nyresvigtet skrider frem, udvikles arteriel hypertension. Hos børn opdages det oftere i ungdomsårene og i ældre aldersgrupper.
Patienter med arvelig nefritis er karakteriseret ved tilstedeværelsen af forskellige (mere end 5-7) stigmaer af bindevævsdysmorfogenese. Blandt bindevævsstigmaerne hos patienter er de mest almindelige hypertelorisme i øjnene, høj gane, bidanomalier, unormal form af ørene, krumning af lillefingeren på hænderne og "sandalgab" på fødderne. Arvelig nefritis er karakteriseret ved ensartetheden af dysmorfogenese-stigmaer inden for en familie, samt en høj frekvens af deres fordeling blandt slægtninge til probander, langs hvis linje sygdommen overføres.
I de tidlige stadier af sygdommen observeres et isoleret fald i partielle nyrefunktioner: transport af aminosyrer, elektrolytter, koncentrationsfunktion, acidogenese, senere ændringer påvirker den funktionelle tilstand af både den proximale og distale del af nefronen og er karakteriseret ved kombinerede partielle lidelser. Et fald i glomerulær filtration forekommer senere, oftere i ungdomsårene. Efterhånden som arvelig nefritis skrider frem, udvikles anæmi.
Således er arvelig nefritis karakteriseret ved et trinvis forløb af sygdommen: først et latent stadium eller skjulte kliniske symptomer, manifesteret ved minimale ændringer i urinsyndromet, derefter forekommer en gradvis dekompensation af processen med et fald i nyrefunktionen med manifeste kliniske symptomer (forgiftning, asteni, udviklingsforsinkelse, anæmi). Kliniske symptomer optræder normalt uanset lagdelingen af den inflammatoriske reaktion.
Arvelig nefritis kan manifestere sig i forskellige aldersperioder, hvilket afhænger af genets virkning, som er i en undertrykt tilstand indtil et bestemt tidspunkt.
Klassifikation
Der er tre typer arvelig nefritis
- Mulighed I - manifesterer sig klinisk som nefritis med hæmaturi, høretab og øjenskade. Nefritis forløber progressivt med udviklingen af kronisk nyresvigt. Arvemåden er dominant, knyttet til X-kromosomet. Morfologisk afsløres en krænkelse af basalmembranens struktur, dens udtynding og opsplitning.
- Mulighed II - manifesterer sig klinisk som nefritis med hæmaturi uden høretab. Nefritis forløb er progressivt med udvikling af kronisk nyresvigt. Arvemåden er dominant, knyttet til X-kromosomet. Morfologisk påvises en udtynding af den glomerulære kapillære basalmembran (især laminadensa).
- Mulighed III - benign familiær hæmaturi. Forløbet er gunstigt, kronisk nyresvigt udvikler sig ikke. Arvetypen er autosomal dominant eller autosomal recessiv. Ved den autosomal recessive arvetype ses et mere alvorligt sygdomsforløb hos kvinder.
Diagnose af Alports syndrom
Følgende kriterier foreslås:
- tilstedeværelsen af mindst to patienter med nefropati i hver familie;
- hæmaturi som det ledende symptom på nefropati hos probanden;
- tilstedeværelsen af høretab hos mindst ét familiemedlem;
- udvikling af kronisk nyresvigt hos en eller flere slægtninge.
I diagnostikken af forskellige arvelige og medfødte sygdomme lægges der stor vægt på en omfattende tilgang til undersøgelsen og frem for alt på at være opmærksom på de data, der opnås ved udarbejdelsen af barnets stamtavle. Diagnosen Alport syndrom anses for gyldig i tilfælde, hvor 3 ud af 4 typiske tegn opdages hos patienten: tilstedeværelsen af hæmaturi og kronisk nyresvigt i familien, tilstedeværelsen af neurosensorisk høretab, synspatologi hos patienten, påvisning af tegn på spaltning af den glomerulære basalmembran med en ændring i dens tykkelse og ujævne konturer under elektronmikroskopiske karakteristika af biopsien.
Patientens undersøgelse bør omfatte kliniske og genetiske forskningsmetoder; målrettet undersøgelse af sygdomshistorien; generel undersøgelse af patienten under hensyntagen til diagnostisk signifikante kriterier. I kompensationsfasen kan patologi kun påvises ved at fokusere på sådanne syndromer som tilstedeværelsen af en arvelig byrde, hypotension, multiple stigmatiseringer af dysembryogenese, ændringer i urinsyndromet. I dekompensationsfasen kan ekstrarenale symptomer forekomme, såsom alvorlig forgiftning, asteni, forsinket fysisk udvikling, anæmi, der manifesterer sig og intensiveres med et gradvist fald i nyrefunktionen. Hos de fleste patienter observeres følgende med et fald i nyrefunktionen: nedsat acido- og aminogenese; 50% af patienterne bemærker et signifikant fald i nyrernes sekretoriske funktion; begrænset udvalg af udsving i urinens optiske tæthed; forstyrrelse af filtreringsrytmen og derefter et fald i glomerulær filtration. Stadiet af kronisk nyresvigt diagnosticeres, når patienter har et forhøjet niveau af urinstof i blodserum (mere end 0,35 g/l) i 3-6 måneder eller mere, og et fald i glomerulær filtration til 25% af normen.
Differentialdiagnostik af arvelig nefritis bør primært udføres ved den hæmaturiske form af erhvervet glomerulonefritis. Erhvervet glomerulonefritis har oftest en akut indsættende periode på 2-3 uger efter en infektion, ekstrarenale tegn, herunder hypertension fra de første dage (ved arvelig nefritis derimod hypotension), nedsat glomerulær filtration ved sygdommens debut, ingen forringelse af partielle tubulære funktioner, hvorimod de er til stede ved arvelig. Erhvervet glomerulonefritis forekommer med mere udtalt hæmaturi og proteinuri, med øget ESR. Typiske ændringer i den glomerulære basalmembran, karakteristiske for arvelig nefritis, er af diagnostisk værdi.
Differentialdiagnostik for dysmetabolisk nefropati udføres ved kronisk nyresvigt, i familien klinisk påviste heterogene nyresygdomme, og der kan være et spektrum af nefropati fra pyelonefritis til urolithiasis. Børn har ofte klager over smerter i maven og periodisk under vandladning, i urinsedimentet - oxalater.
Ved mistanke om arvelig nefritis bør patienten henvises til en specialiseret nefrologisk afdeling for at afklare diagnosen.
Hvad skal man undersøge?
Hvordan man undersøger?
Hvilke tests er nødvendige?
Hvem skal kontakte?
Behandling af Alports syndrom
Kuren omfatter begrænsninger af hård fysisk anstrengelse og udsættelse for frisk luft. Kosten er komplet med tilstrækkelige niveauer af komplette proteiner, fedtstoffer og kulhydrater, under hensyntagen til nyrefunktionen. Af stor betydning er påvisning og behandling af kroniske infektionsfokus. Følgende lægemidler anvendes: ATP, cocarboxylase, pyridoxin (op til 50 mg/dag), carnitinchlorid. Kurserne administreres 2-3 gange om året. Ved hæmaturi ordineres urtemedicin - brændenælde, aroniasaft, røllike.
Der findes rapporter i udenlandsk og indenlandsk litteratur om behandling med prednisolon og brug af cytostatika. Det er dog vanskeligt at bedømme effekten.
Ved kronisk nyresvigt anvendes hæmodialyse og nyretransplantation.
Der findes ingen metoder til specifik (effektiv patogenetisk) terapi for arvelig nefritis. Alle behandlingsforanstaltninger sigter mod at forebygge og bremse nedgangen i nyrefunktionen.
Kosten bør være afbalanceret og kalorierig, idet der tages hensyn til nyrernes funktionelle tilstand. I mangel af funktionelle forstyrrelser bør barnets kost indeholde tilstrækkeligt med proteiner, fedtstoffer og kulhydrater. Ved tegn på nyredysfunktion bør mængden af protein, kulhydrater, calcium og fosfor begrænses, hvilket forsinker udviklingen af kronisk nyresvigt.
Fysisk aktivitet bør begrænses; børn rådes til at undgå sport.
Kontakt med smitsomme patienter bør undgås, og risikoen for udvikling af akutte luftvejssygdomme bør reduceres. Hygiejne ved foci af kronisk infektion er nødvendig. Forebyggende vaccinationer udføres ikke for børn med arvelig nefritis, vaccination er kun mulig ved epidemiologiske indikationer.
Hormonal og immunsuppressiv behandling ved arvelig nefritis er ineffektiv. Der er indikationer på en vis positiv effekt (reduktion af proteinuri og bremsning af sygdomsprogression) ved langvarig flerårig brug af cyclosporin A og ACE-hæmmere.
Ved behandling af patienter anvendes lægemidler, der forbedrer stofskiftet:
- pyridoxin - 2-3 mg/kg/dag i 3 doser i 4 uger;
- cocarboxylase - 50 mg intramuskulært hver anden dag, i alt 10-15 injektioner;
- ATP - 1 ml intramuskulært hver anden dag, 10-15 injektioner;
- A-vitamin - 1000 IE/år/dag i 1 dosis i 2 uger;
- E-vitamin - 1 mg/kg/dag i 1 dosis i 2 uger.
Denne type terapi hjælper med at forbedre patienternes generelle tilstand, reducere tubulære dysfunktioner og udføres i forløb 3 gange om året.
Levamisol kan anvendes som en immunmodulator - 2 mg/kg/dag 2-3 gange om ugen med pauser mellem doserne på 3-4 dage.
Ifølge forskningsdata har hyperbarisk iltning en positiv effekt på sværhedsgraden af hæmaturi og nyredysfunktion.
Den mest effektive metode til behandling af arvelig nefritis er rettidig nyretransplantation. I dette tilfælde er der intet tilbagefald af sygdommen under transplantationen; i en lille procentdel af tilfældene (ca. 5%) kan nefritis udvikles i den transplanterede nyre forbundet med antigener til den glomerulære basalmembran.
En lovende retning er prænatal diagnostik og genteknologisk behandling. Dyreforsøg viser høj effektivitet i at overføre normale gener, der er ansvarlige for syntesen af type IV kollagen alfakæder, til nyrevæv, hvorefter syntesen af normale kollagenstrukturer observeres.
Vejrudsigt
Prognosen for arvelig nefritis er altid alvorlig.
Prognostisk ugunstige kriterier for forløbet af arvelig nefritis er:
- mandligt køn;
- tidlig udvikling af kronisk nyresvigt hos familiemedlemmer;
- proteinuri (mere end 1 g/dag);
- fortykkelse af de glomerulære basalmembraner ifølge mikroskopi;
- akustisk neuritis;
- deletion i Col4A5-genet.
Prognosen for benign familiær hæmaturi er mere gunstig.
Использованная литература