Medicinsk ekspert af artiklen

Nye publikationer
Mucopolysaccharidosis type 3
Sidst revideret: 23.04.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
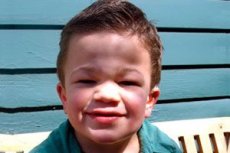
Mucopolysaccharidose type III (synonymer: Sanfilippo syndrom, manglende lysosomal aN-acetylglucosaminidase - mucopolysaccharidosis Sh A, acetyl-CoA og-glucosaminid-N-acetyltransferase - mukopolysakkaridose III B, N-acetylglucosamin-6-sulfatase - mukopolysakkaridose III C, sulfamidazy - mucopolysaccharidose III-D).
 [
[Patogenese
Ved sygdom forårsager mutationer fire forskellige gener: lysosomal aN-acetylglucosaminidase (mucopolysaccharidosis III A), acetyl-CoA og-glucosaminid-N-acetyltransferase (mucopolysaccharidosis III B), lysosomal N-acetylglucosamin-6-sulfatase (mucopolysaccharidosis III C) sulfamidazy (mucopolysaccharidosis III D). Alle enzymer er involveret i metabolisme af heparansulfat.
Heparan-N-sulfatase-genet-SGSH-er placeret på den lange arm af kromosomet 17-17q25,3. 75,3% af de aktuelt kendte mutationer i SGSH-genet er punktmutationer. Hyppige mutationer, der er typiske for europæiske befolkninger - R74C (56% i Polen og 21% i Tyskland) og R245H (56% i Holland) er beskrevet.
Mutationsfrekvensen for R74C er 47,5%, mutationen af R245H er 7,5%. De to andre mutationer, delll35G og N389S, udgør sammen 21,7% af de mutante alleler.
AN-acetyl-glucosaminidase-genet, NAGLU, er placeret på den lange arm af kromosomet 17-17q21. 69% af mutationerne fundet i NAGLU genet er missense og nonsensmutationer, 26,3% er små deletioner og insertioner. Acetyl-CoA-cc-glucosaminid-N-acetyltransferase-genet, HGSNAT, er placeret på den korte arm af kromosomet 8-8p11.1. Genet blev karakteriseret kun i 2006 og til dato fandt det kun få mutationer.
Genet N-acetyl-glucosamin-6-sulfatase - GNS - er placeret på den lange arm af kromosom 12-12ql4. Der er 12 patienter med mucopolysaccharidose IIID registreret i verden. Der er 4 mutationer i GNS genet.
Når alle undertyper III mucopolysaccharidosis overtrædelsen sker nedbrydning af heparansulfat, indgår i strukturen af cellemembraner, herunder membraner af neuroner, som korrelerer med alvorlige neurodegenerative processer forårsaget af kortikal atrofi. Kronisk diarré forklares ved involvering i det autonome nervesystems patologiske proces sammen med dysfunktion i tarmslimhinden. Sensorineuralt høretab skyldes sandsynligvis tre årsager: hyppig otitis, deformationer af de ørehæmmende øjne og anomalier i det indre øre. Stivheden af leddene er resultatet af deformationen af metafyserne, fortykkelsen af ledkapslen er sekundær til aflejringen af glycosaminoglycaner og fibrose i den. Intrasyndromiske forskelle i sygdommens sværhedsgrad skyldes udelukkende den resterende funktionelle aktivitet af det mutante enzym: Jo højere det er, jo lettere sygdommen skrider frem.
Symptomer mucopolysaccharidosis type 3
Klinisk polymorfisme i Sanfilippos syndrom er mindre udtalt end i andre typer af mucopolysaccharidose. Karakteriseret af en langsom udvikling af sygdommen, alvorlige neurologiske lidelser med svære symptomer fra de indre organer og andre systemer.
De første symptomer på sygdommen opstår normalt i en alder af 2-6 år hos børn med en tidligere normal udvikling. Manifest symptomer omfatter regression af psykomotorisk og taleudvikling, psykiatriske lidelser i form af udvikling af hyperaktivitetssyndrom, autistisk eller aggressiv adfærd, søvnforstyrrelser; børn bliver skødesløse og uopmærksom.
Andre almindelige symptomer er hirsutisme, hårdt hår, mild hepatosplenomegali, valgus deformitet af ekstremiteterne, kort hals. Dannelsen af grove ansigtstræk efter type gargoilizma og skeletal deformitet multipel dysostosis svagt udtrykt ved Mucopolysaccharidosis III sammenlignet med andre typer af mucopolysaccharidosis, kendetegnet Hurler fænotype. Væksten svarer som regel til alder, og fælles stivhed forårsager sjældent en krænkelse af deres funktioner. De fleste patienter udvikler ofte osteoporose og osteomalaci. Sekundære skeletforstyrrelser er en høj risiko for patologiske frakturer. Grove psykoneurologiske lidelser observeres hyppigst i det 6-10 år i livet, de fører til alvorlig social fejlpasning. Progressivt neurosensorisk høretab er iboende hos alle patienter med svære og moderate former for sygdommen. Beslaglæggelser observeres hos næsten alle patienter som sygdommen skrider frem.
Sygdommens forløb udvikler sig hurtigt, de fleste patienter overlever ikke i alderen 20 år. Det antages, at mucopolysaccharidosis IIIA er den mest almindelige og alvorlige type af dette syndrom.
Diagnosticering mucopolysaccharidosis type 3
Diagnosen af mucopolysaccharidose III bekræftes ved at bestemme niveauet for uringlucosamycin udskillelse og måling af enzymaktivitet. I tilfælde af mucopolysaccharidose III øges den totale udskillelse af glycosaminoglycaner i urinen, og hyperexcretionen af heparansulfat observeres. Aktiviteten af lysosom enzymer svarende til en bestemt subtype af mucopolysaccharidosis III måles i leukocytter eller en kultur af hudfibroblaster under anvendelse af et kunstigt fluorogent substrat.
Prænatal diagnose er mulig ved måling enzymaktivitet i moderkagebiopsi ved 9-11 ugers svangerskab og / eller bestemmelse af spektret af glycosaminoglycaner i fostervandet på 20-22 uger af graviditeten. For familier med en kendt genotype er det muligt at udføre DNA-diagnostik i de tidlige stadier af graviditeten.
Hvilke tests er nødvendige?
Differential diagnose
Differentialdiagnosticering sker inden Mucopolysaccharidose gruppe, og med andre lysosomale aflejringssygdomme: Mucolipidosis, galaktosialidozom, sialidosis, mannozidozom, Fucosidosis, GM1-gangliosidose.
Hvem skal kontakte?
Behandling mucopolysaccharidosis type 3
Hidtil er effektive terapier til mucopolysaccharidose III ikke blevet udviklet. Symptomatisk terapi er indiceret.
Использованная литература