Medicinsk ekspert af artiklen

Nye publikationer
Subakut nekrotiserende Leahs encefalomyopati
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
 [
[ Årsager af Leahs syndrom
Sygdommen er baseret på en mangel på enzymer, der sørger for energiproduktion, primært på grund af en forstyrrelse af pyruvinsyremetabolismen og en defekt i elektrontransporten i respirationskæden. Der udvikles en mangel på pyruvatdehydrogenasekomplekset (a-E1-underenhed), pyruvatcarboxylase, kompleks 1 (NAD-coenzym Q-reduktase) og kompleks 4 (cytochromoxidase) i respirationskæden.
Det er blevet fastslået, at defekter i pyruvatcarboxylase, kompleks 1 (NAD-coenzym Q-reduktase) og kompleks 4 (cytochromoxidase) i respirationskæden nedarves autosomalt recessivt, mens defekter i pyruvatdehydrogenasekomplekset (a-E1-underenhed) nedarves X-bundet recessivt. Ved punktmutationer i mtDNA, som påvirker den 6. underenhed af ATPase, er mitokondrienedarvning typisk. Oftest forekommer en miscens-mutation, der er forbundet med erstatning af thymin med guanin eller cytosin i position 8993 af mtDNA. Mindre almindelig er en mutation i position 9176 af mtDNA. Da T8993G-mutationen er den primære defekt i NARP-syndrom, er familier med disse to sygdomme blevet beskrevet. Hos børn er der også beskrevet en mutation i mtDNA i position 8344, som forekommer ved MERRF-syndrom.
Det antages, at der i tilfælde af ophobning af mutant mtDNA i de fleste mitokondrier udvikles et alvorligt forløb af Leigh syndrom. Ved den mitokondrielle oprindelse af denne tilstand findes mutant mtDNA i 90% af alle mitokondrier. Patogenesen er forbundet med en forstyrrelse af energidannelsen i cellerne og udviklingen af mælkesyreacidose.
Symptomer af Leahs syndrom
De første tegn på sygdommen debuterer i en tidlig alder (1-3 år). Der er dog kendte tilfælde af sygdomsmanifestation i 2-ugers alderen og i 6-7-årsalderen. I starten udvikles uspecifikke lidelser: forsinket psykomotorisk udvikling, nedsat appetit, opkastningsepisoder, vægttab. Efterfølgende øges neurologiske symptomer: muskelhypotoni eller dystoni med overgang til hypertoni, myokloniske anfald eller tonisk-kloniske anfald, tremor i ekstremiteterne, koreoathetose, koordinationsforstyrrelser, nedsatte senereflekser, sløvhed, døsighed. Cerebral neurodegeneration er progressiv. Symptomer på pyramideformet og ekstrapyramidal insufficiens øges, synkeevnen er nedsat. Sådanne ændringer i synsorganet som ptose, oftalmoplegi, atrofi af synsnerverne, sjældnere pigmentdegeneration af nethinden observeres ofte. Nogle gange udvikles hypertrofisk kardiomyopati, episoder med takypnø optræder.
I sjældne tilfælde forløber sygdommen som akut encefalopati. Mere typisk er et kronisk eller subakut forløb, som fører til en dødelig udgang flere år efter sygdommens debut. Ved et hurtigt forløb (flere uger) indtræffer døden som følge af lammelse af åndedrætscentret.
Diagnosticering af Leahs syndrom
En biokemisk blodprøve afslører mælkesyreacidose på grund af ophobning af mælkesyre og pyrodruesyre i blodet og cerebrospinalvæsken, samt en stigning i alaninindholdet i blodet. Niveauet af ketonstoffer kan også være forhøjet. Øget udskillelse af organiske syrer påvises i urinen: mælkesyre, fumarsyre osv. Niveauet af carnitin i blodet og vævet falder ofte.
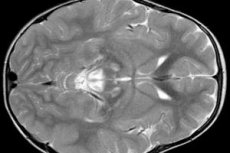
EEG-resultater afslører fokale tegn på epileptisk aktivitet. MR-data afslører forstørrelse af hjerneventriklerne, bilateral hjerneskade, forkalkning af basalganglierne (nucleus caudatus, putamen, substantia nigra, globus pallidus). Atrofi af hjernehalvdelene og hjernesubstansen kan også detekteres.
Morfologisk undersøgelse afslører store ændringer i hjernens substans: symmetriske nekrosefokus, demyelinisering og svampet degeneration af hjernen, primært i de midterste sektioner, pons, basalganglier, thalamus og synsnerven. Det histologiske billede inkluderer cystisk degeneration af hjernevæv, astrocytisk gliose, neuronal død og en stigning i antallet af mitokondrier i cellerne. I skeletmuskulaturen ses ophobning af lipidindeklusioner, et fald i den histokemiske reaktion på komplekserne 1 og 4 i respirationskæden, subsarkolemmal ophobning af mitokondrier, unormale mitokondrier med desorganisering af cristae. RRF-fænomenet detekteres ofte ikke.
Hvordan man undersøger?
Hvilke tests er nødvendige?
Использованная литература