Medicinsk ekspert af artiklen

Nye publikationer
Xeroderma pigmentosum
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
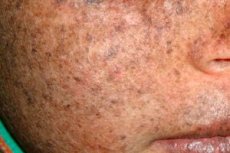
Xeroderma pigmentosum er en autosomal recessiv genetisk lidelse i DNA-reparation. Sygdommen er forårsaget af mutationer i gener, der er involveret i reparation af DNA beskadiget af ultraviolet stråling og andre giftige stoffer.
Oftest rammer denne sygdom børn, som også kaldes "nattens børn". Hyppige komplikationer af sygdommen er basalcellekarcinom og andre maligne neoplasmer i huden, metastatisk malignt melanom og pladecellekarcinom.
 [ 1 ]
[ 1 ]
Patogenese
Mangel på UV-endonukleaseenzymer i patientens celler (i fibroblaster) eller deres fuldstændige fravær spiller en vigtig rolle i sygdommens udvikling. Dette enzym er ansvarligt for reproduktionen af DNA, der er beskadiget af ultraviolette stråler. Ifølge andre kilder spiller mangel på DNA-polymerase-1-enzymet en vigtig rolle i sygdommens udvikling hos nogle patienter. Sygdommen udvikler sig oftest under påvirkning af stråler med en bølgelængde på 280-310 nm. Det menes, at pigmentxerodermi udvikles på grund af indtrængen af forskellige fotodynamiske stoffer i kroppen eller en stigning i porphyriner i det menneskelige biologiske miljø.
I de tidlige stadier af sygdommen observeres hyperkeratose, atrofi af epidermisfokus, udtynding af Malpighi-laget, en stigning i melaningranulat i det basale cellelag og kronisk inflammation, der infiltrerer i det øvre lag af dermis (hovedsageligt omkring blodkar). Efterfølgende opdages degenerative forandringer i kollagen og elastiske fibre, og i tumorstadiet afsløres tegn, der er karakteristiske for hudkræft.
Symptomer xeroderma pigmentosum
Mænd og kvinder er ligeligt berørt af denne sygdom. Sygdommen begynder i den tidlige barndom om foråret eller sommeren. Patientens hud er særligt følsom over for sollys. Derfor optræder det første udslæt i form af hyperpigmentering, der ligner en modermærke, på områder af huden, der er udsat for sollys. Udslættet forværres, erytem intensiveres, bliver mørkebrunt, telangiektasi, angiom og atrofi optræder. Derefter vokser papillomer og vorter (hovedsageligt på huden i ansigtet og på halsen), pletter og sår opstår. Som følge af sårdannelse bliver næsen tyndere ("fuglenæb"), og øjenlåget krænger ud. Papillomer udvikler sig normalt til ondartede tumorer. Pigmenteret xerodermi optræder som regel sammen med spinocellulær kræft, melanosarkomer.
Hvad generer dig?
Niveauer
I det kliniske forløb af xeroderma pigmentosum skelnes der mellem fem stadier: inflammatorisk (erythematøs), hyperpigmentering, atrofi, hyperkeratose og maligne tumorer.
Under den inflammatoriske (erytematøse) fase opstår der hævelse, røde pletter og undertiden blærer og vesikler på hudområder, der er udsat for sollys (ansigt, hals, øvre del af brystet, arme, hænder). Der er næsten intet udslæt på områder, der ikke er udsat for sollys.
Ved hyperpigmentering opstår der lysebrune, brune hyperpigmenterede pletter, der ligner modermærker, i stedet for de røde pletter.
Ved atrofi tørrer huden ud, bliver tyndere, og der dannes rynker. Talrige små, stjerneformede telangiektasier og ar med en skinnende overflade observeres på huden på læber og næse. På grund af atrofi og ar udvikles mikrotomi (reduktion af mundåbningen), ektropion, udtynding af ører og næse, atresi af næseåbningen og munden. Hos 80-85% af patienterne er øjnene påvirket: konjunktivitis, keratitis, skader på hornhinden og slimhinderne samt svækkelse af synet observeres. Dyskromi, telangiektasi, hyperkeratotiske forandringer og tumorer observeres på huden på øjenlågene.
Ved hyperkeratose forekommer vortede tumorer, papillomer, keratoacanthomer, fibromer, angiofibromer og andre godartede tumorer i de ovenfor beskrevne patologiske foci. Derfor inkluderer nogle forskere pigmentxerodermi i gruppen af præcancerøse sygdomme.
Efter 10-15 år fra sygdommens debut opstår maligne hudtumorer (basaliom, endoteliom, angiosarkom) i atrofisk ændrede foci og pigmentpletter. Tumorerne ødelægges på kort tid, metastaserer til indre organer og fører til døden. I de indre organer og væv hos nogle patienter observeres generelle dystrofiske forandringer (syndaktelier i anden og tredje tå, tanddystrofi, fuldstændigt hårtab osv.).
Forms
Den neurologiske form af xeroderma pigmentosum manifesterer sig i to syndromer.
Reed syndrom er karakteriseret ved kliniske manifestationer af xeroderma pigmentosum og mikrocefali, idiopati og langsom vækst af skeletsystemet. I sygdommens neurologiske form er DNA-reparation vanskelig, og røntgenbehandling fører til en stigning i de primære patologiske foci.
De Sinctis-X Cocchione syndrom er karakteriseret ved følgende kliniske tegn:
- udvikling af kliniske tegn på xeroderma pigmentosum på særligt lysfølsom hud;
- tidlig forekomst af ondartede tumorer;
- samtidig forløb af spastisk lammelse, mikrocefali og demens;
- medfødte deformiteter og dværgvækst;
- underudvikling af gonaderne;
- hyppige aborter;
- recessiv arvelighed.
Ifølge nogle dermatologer er Santis-Cacchione syndrom ikke en uafhængig sygdom, men en alvorlig og fuldt ud udtrykt klinisk manifestation af xeroderma pigmentosum.
Genetiske former
Typer |
Gen |
Lokus |
Beskrivelse |
Type A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) gruppe A – klassisk form |
Type B, II, XPB |
XPB |
2q21 |
XP Gruppe B |
Type C, III, XPC |
XPC |
3p25 |
XP Gruppe C |
Type D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP gruppe D eller De Sanctis-Cacchione syndrom |
Type E, V, XPE |
DDB2 |
23:00-23:00 |
XP Gruppe E |
Type F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Gruppe F |
Type G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP gruppe G og COFS syndrom (cerebro-oculo-facioskeletalt syndrom) type 3 |
Type V, XPV |
POLH |
6p21.1-p12 |
Variant Xeroderma Pigmentosa |
Hvad skal man undersøge?
Hvordan man undersøger?
Hvem skal kontakte?
Behandling xeroderma pigmentosum
Malariamidler (delagyl, plaquenil, resoquin osv.), der beskytter DNA mod ultraviolette stråler, hæmmer depolymeriseringsprocessen og reducerer hudens følsomhed (fotosensibilisering) over for sollys. Disse lægemidler har antiinflammatoriske og hyposensibiliserende egenskaber. Det anbefales at udføre generel behandling sammen med vitaminbehandling (B1, B2, PP, B6, B12, A, E), antihistaminer (tavegil, diphenhydramin, suprastin) og desensibiliserende midler (natriumthiosulfat, 10% calciumchlorid intravenøst 10 ml).
Til lokal behandling anvendes solcremer og salver.
Ved tumorformen af pigmentxerodermi anvendes kirurgiske metoder, flydende nitrogen og laserstråler. For at beskytte dig mod sollys skal du bære løstsiddende tøj, solhatte og handsker.
Vejrudsigt
De fleste patienter (2/3) dør, før de fylder 15 år. Nogle patienter kan leve i 40-50 år.
[ 20 ]