Medicinsk ekspert af artiklen

Nye publikationer
Angelmans syndrom hos børn og voksne
Sidst revideret: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.

Der findes en række sygdomme, hvor udtryk som "pas på dig selv, så bliver du ikke syg" lyder i det mindste latterlige. Det er patologier, hvor visse mentale og fysiske abnormiteter er iboende i barnets krop allerede før fødslen, men forældrene er ikke skyld i dette. Sådanne sygdomme er forårsaget af mutationer eller abnormiteter i kromosomsæt og kaldes kromosomale eller genetiske. Angelman syndrom, Downs syndrom, Patau syndrom, Edwards syndrom, Turner syndrom, Prader-Willi syndrom - dette er kun en del af de genetiske sygdomme fra en ret anstændig liste.
Glad mand-syndrom
Denne gang vil vi tale om patologien, der er opkaldt efter den engelske børnelæge Harry Angelman, som først rejste spørgsmålet om dette problem i 1965, efter at have stødt på tre usædvanlige børn i sin praksis dagen før, forenet af fælles, ejendommelige symptomer. Lægen kaldte disse børn for dukkebørn og skrev en artikel om dem, der oprindeligt hed "Børn-marionetter". Selve artiklen og dens titel blev skrevet under indtryk af et maleri, der var set på et af museerne i Verona. Maleriet forestillede en grinende dreng, og det blev kaldt "Dukkedrengen". Forbindelsen mellem barnet, der er afbildet på maleriet, og de tre børn, som Angelman engang mødte i sin praksis, fik børnelægen til at samle børnene i én gruppe på grund af den sygdom, de havde.
Der er intet overraskende i, at de børn, der er nævnt i artiklen, ikke blev bemærket af andre læger. Ved første øjekast så det jo ud til, at de havde helt forskellige sygdomme, så forskelligt var det generelle kliniske billede af sygdommen i 3 forskellige tilfælde. Måske ville den "nye" kromosomale patologi have interesseret andre forskere, men på det tidspunkt var genetikken endnu ikke udviklet nok til at bekræfte den engelske læges hypotese. Derfor blev artiklen, efter en vis interesse for den, smidt på baghylden i lang tid.
Den næste omtale af Angelman syndrom, som artiklen af den engelske børnelæge G. Angelman nu hed, stammer fra begyndelsen af 80'erne i det 20. århundrede. Og først i 1987 var det muligt at finde årsagen til, at en lille del af børnene fødes med sådanne afvigelser, at de udefra ser ud til at være konstant smilende og glade. Faktisk er dette slet ikke sandt, og smilet er blot en grimasse, bag hvilken der gemmer sig en ulykkelig menneskesjæl og forældrenes smerte.
Epidemiologi
Ifølge statistikker kan en kromosomal mutation hos et barn udvikle sig både på baggrund af lignende mutationer hos forældrene og i fravær af sådanne. Der er ingen klar arvelig karakter af Angelman syndrom (AS), men sandsynligheden for at udvikle patologi hos forældre med kromosomale mutationer er ret høj.
Det er også interessant, at hvis en familie allerede har et barn med AS, er der en procents chance for at få et andet barn med samme lidelse, selvom forældrene er raske.
Der findes stadig ingen præcis statistik over antallet af patienter med Angelmans syndrom. Måske skyldes det de mange forskellige symptomer, som kan forekomme i en bestemt sammensætning eller slet ikke forekomme i lang tid. Det antages, at sygdommens prævalens er: 1 barn pr. 20.000 nyfødte. Men dette tal er meget omtrentligt.
Årsager Angelmans syndrom
Angelman syndrom er et medicinsk navn for en kromosomal patologi, men det er langt fra det eneste. Folk kalder denne sygdom for dukkebørnesyndrom, gladdukkesyndrom, Petrushka syndrom og grinende dukkesyndrom. Folk finder på alle mulige navne (nogle gange endda stødende for patienterne selv og deres forældre), men en sygdom er en sygdom, uanset hvor sjov den ser ud, og uanset hvad årsagerne er.
Og årsagerne til udviklingen af Angelman syndrom, ligesom mange andre genetiske patologier, er i alle tilfælde forstyrrelser i strukturen af et af kromosomerne eller kromosomsættet som helhed. Men i vores tilfælde ligger hele problemet i kromosom 15, som er arvet fra moderen. Det vil sige, at faderkromosomet i dette tilfælde ikke har nogen afvigelser, men det kvindelige kromosom gennemgår visse mutationer.
Ifølge typen af kromosomafvigelse klassificeres Angelmans syndrom som en kromosomal mutation. Sådanne mutationer betragtes som:
- En deletion (fravær af en del af et kromosom, der indeholder et bestemt sæt gener; hvis et af generne mangler, taler vi om en mikrodeletion), som er resultatet af to brud og én genforening, hvor en del af det oprindelige kromosom går tabt.
- Duplikation (tilstedeværelsen af en ekstra sektion i et kromosom, der er en kopi af en eksisterende), hvilket i de fleste tilfælde fører til en persons død og sjældnere til infertilitet.
- Inversion (omvending af en af kromosomets sektioner med 180 grader, dvs. i den modsatte retning, og så er generne i den placeret i den modsatte rækkefølge), når de knækkede ender af kromosomet er forbundet i en anden rækkefølge end den oprindelige.
- Indsættelse (hvis en del af det genetiske materiale i et kromosom er forkert placeret),
- translokation (hvis en bestemt del af et kromosom er bundet til et andet kromosom; en sådan mutation kan være gensidig uden tab af sektioner).
Hvis barnet modtager et muteret kromosom fra en intetanende mor, er det dømt til at blive født med abnormiteter. Den mest almindelige årsag til Angelmans syndrom anses stadig for at være en deletion af moderens 15. kromosom, når en lille del mangler. Mindre almindelige mutationer i "latterdukke"-syndromet anses for at være:
- translokation,
- unipaternal disomi (hvis barnet har modtaget et par kromosomer fra faderen, mangler moderens kromosom)
- mutation af gener i DNA, som både er det primære byggemateriale (genetisk materiale) og instruktioner til dets korrekte anvendelse (især mutation af ube3a-genet i moderens kromosom).
Tilstedeværelsen af en af disse mutationer hos forældre er en risikofaktor for udvikling af Angelmans syndrom hos børn. Men ikke kun kromosomale mutationer, men også genomiske (som er forbundet med en kvantitativ ændring i kromosomsæt og er mere almindelige end kromosomale) kan provokere udviklingen af sygdommen hos et barn. Almindelige genomiske mutationer omfatter kromosomtrisomi (hvis en persons kromosomsæt har mere end 46 kromosomer).
For at en patologi kan opstå hos et barn, er det slet ikke nødvendigt, at forældrene har kromosomafvigelser. Og alligevel er der en vis procentdel af patienter, hvis sygdom er arvelig.
Patogenese
Lad os dykke lidt dybere ned i biologi, eller mere præcist, genetik. Den genetiske information om hver enkelt menneskelig organisme er indeholdt i 23 par kromosomer. Et kromosom fra et par gives videre til barnet fra faderen, det andet fra moderen. Alle kromosompar er forskellige i form og størrelse og bærer bestemte oplysninger. Således er det 23. par kromosomer (X- og Y-kromosomer) ansvarligt for dannelsen af barnets seksuelle karakteristika (XX - pige, XY - dreng, mens Y-kromosomet kun kan modtages af barnet fra faderen).
Ideelt set modtager et barn 46 kromosomer fra sine forældre, som danner barnets genetiske karakteristika og forudbestemmer det som individ. Et større antal kromosomer kaldes trisomi og betragtes som en afvigelse fra normen. For eksempel forårsager tilstedeværelsen af kromosom 47 i kromosomsættet (karyotype, artsbestemmende og individuelle karakteristika) forekomsten af Downs syndrom.
Hvis kromosomerne farves med et specielt farvestof, kan man under mikroskopet se striber i forskellige nuancer langs hver af dem. Inde i hver stribe er der et stort antal gener. Alle disse striber er nummererede af forskere og har en fast placering. Fraværet af en af striberne betragtes som en afvigelse fra normen. Ved Angelman syndrom kan man meget ofte observere fraværet af segmenter af moderens kromosom i intervallet q11-q13, placeret i den lange arm, hvor antallet af DNA-baser kun er omkring 4 millioner.
Hovedkomponenten i kromosomet anses for at være et utrolig langt DNA-molekyle, der indeholder tusindvis af gener og titusinder og hundredvis af millioner nitrogenholdige baser. Således indeholder kromosom 15, der er ansvarlig for udviklingen af Angelman syndrom og flere andre, 1200 gener og omkring 100 millioner baser. Enhver forstyrrelse i DNA-molekylets struktur vil helt sikkert påvirke det kommende barns udseende og udvikling.
Den genetiske information indeholdt i gener omdannes til protein eller RNA. Denne proces kaldes genekspression. På denne måde får den genetiske information, der modtages fra forældre, både form og indhold, hvilket er legemliggjort i deres unikke kvindelige eller mandlige arving.
Der findes en række patologier med en ikke-klassisk arvstype, herunder Angelman syndrom, hvor gener modtaget fra forældre som en del af parrede kromosomer bærer et unikt præg af forældrene og manifesterer sig på forskellige måder.
Så Angelman syndrom er et slående eksempel på genomisk prægning, hvor genekspression i barnets krop er direkte afhængig af, hvilken forælder allelerne blev modtaget fra (forskellige former af samme gen, modtaget fra far og mor, placeret på identiske sektioner af parrede kromosomer). Det vil sige, at kun anomalier i moderens kromosom fører til udviklingen af syndromet, mens mutationer og strukturelle lidelser i faderens kromosom forårsager helt forskellige patologier.
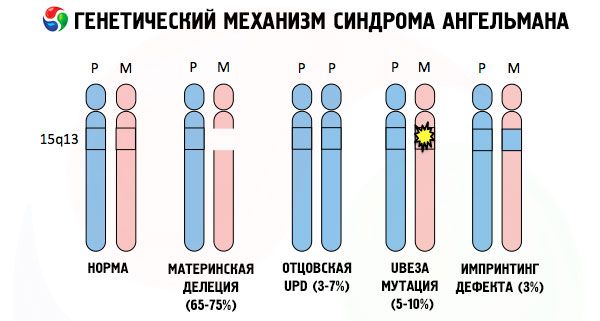
Ved denne patologi er der mangel på bestemte gener i moderens kromosom eller et tab/reduktion i aktiviteten af individuelle gener (i langt de fleste tilfælde ube3a-genet, som er involveret i metabolismen af ubiquitin, et protein, der regulerer nedbrydningen af andre proteiner). Som følge heraf diagnosticeres barnet med mentale udviklingsabnormaliteter og fysiske deformiteter.
Symptomer Angelmans syndrom
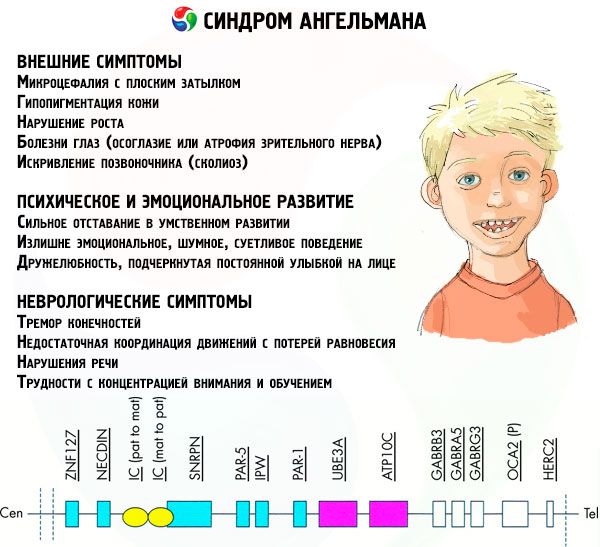
Symptomerne på Angelman syndrom påvirker forskellige aspekter af et barns liv og udvikling: fysiske, neurologiske, mentale. Baseret på dette kan der identificeres 3 grupper af symptomer, der indikerer udviklingen af denne patologi.
- Eksterne eller fysiske symptomer:
- et uforholdsmæssigt lille hoved sammenlignet med kroppen og lemmerne, som er af normal størrelse,
- for bred mund,
- der er næsten altid et smil på læben (med åben mund),
- sparsomme tænder,
- smal overlæbe,
- ofte fremstående bred tunge,
- fremstående underkæbe,
- spids hage,
- meget lys hud, ofte hår (albinisme, forbundet med at kroppen ikke producerer pigmentet melanin),
- mørke pletter på lys hud (hypopigmentering på grund af utilstrækkelig melaninproduktion)
- fysiske eller ydre symptomer: øjensygdomme såsom strabismus eller synsnerveatrofi,
- krumning af rygsøjlen (skoliose),
- stive ben (når man går, bøjer en person ikke benene ved knæene på grund af lav mobilitet i leddene, deraf sammenligningen med en dukkegang).
- Symptomer relateret til mental og følelsesmæssig udvikling:
- svær mental retardering,
- overdrevent følelsesladet, støjende, kræsen adfærd,
- hyppig håndklappning,
- udtrykt venlighed, understreget af et konstant smil på læben,
- hyppig latter uden grund.
- Neurologiske symptomer:
- rystelser i lemmerne,
- utilstrækkelig koordinering af bevægelser med tab af balance,
- nedsat muskeltonus,
- forskellige søvnforstyrrelser,
- hyppige hysteriske anfald i barndommen,
- taleforstyrrelser (barnet begynder at tale sent, har dårlige kommunikationsevner og sløret tale),
- hyperaktivitet på baggrund af øget excitabilitet,
- vanskeligheder med at koncentrere sig og lære.
Men dette er et generaliseret billede af sygdommen. Faktisk afhænger det kliniske billede af Angelman syndrom i høj grad af sygdommens udviklingsstadium og typen af kromosomal mutation, der forårsagede patologien. Det betyder, at sygdommens symptomer kan variere betydeligt hos forskellige patienter, hvilket i lang tid ikke tillod os at skelne patologien fra andre med et lignende klinisk billede.
Blandt det samlede antal symptomer kan vi fremhæve dem, der er karakteristiske for alle patienter uden undtagelse:
- svær mental retardering,
- upassende adfærd (urimelig latter, øget ophidselse, dårlig koncentration, eufori)
- underudvikling af motoriske færdigheder,
- dårlig koordination af bevægelser, gangart ataksi (ujævnt tempo, svajning fra side til side osv.), tremor i lemmerne.
- taleudviklingsforstyrrelse med en overvægt af nonverbale kommunikationsmidler.
Blandt de symptomer, som langt de fleste patienter oplever, kan følgende skelnes:
- uforhold mellem hoved og krop forårsaget af forsinket fysisk udvikling,
- hos mange patienter er kraniets form sådan, at hjernens størrelse forbliver mindre end hos raske mennesker (mikrocefali),
- epileptiske anfald før 3-årsalderen med et progressivt fald i styrke og hyppighed i en ældre alder,
- forvrængning af EEG-parametre (fluktuationer og høj amplitude af lavfrekvente bølger).
Disse symptomer er ret almindelige, men 20% af patienter med Angelman syndrom har dem ikke.
Endnu sjældnere er det muligt at diagnosticere sådanne manifestationer af sygdommen som:
- svær eller mild strabismus,
- dårlig kontrol over tungens bevægelser, hvilket resulterer i, at patienter ofte stikker tungen ud uden grund,
- vanskeligheder med at synke og suge, især hos små børn,
- forstyrrelse af hud- og øjenpigmentering,
- armene hævet eller bøjet under gang,
- hyperrefleksi,
- søvnforstyrrelser, især i barndommen,
- hyppig spytproduktion,
- umættelig tørst,
- overaktive tyggebevægelser,
- overfølsomhed over for varme,
- flad bagside af hovedet,
- fremstående underkæbe,
- glatte håndflader.
En ret stor procentdel af patienterne har problemer med vandladning, som de dårligt kontrollerer, nedsat finmotorik, hvilket skaber vanskeligheder med egenomsorg og indlæring, og overvægt. Næsten alle patienter oplever puberteten senere end raske jævnaldrende.
Børn med Angelman syndrom opfatter og forstår mundtlig tale godt, men ønsker ikke at deltage i samtaler og begrænser deres tale til flere dusin ord, der er nødvendige i hverdagen. I voksenalderen ser sådanne patienter dog yngre ud end deres jævnaldrende uden genetiske patologier.
Mange symptomer på Angelman syndrom er inkonstante, så det kliniske billede af sygdommen ændrer sig betydeligt med alderen. Krampeanfald og epileptiske anfald bliver sjældnere eller forsvinder helt, patienten bliver mindre ophidset, og søvnen forbedres.
Komplikationer og konsekvenser
Angelmans syndrom er en alvorlig, i øjeblikket praktisk talt uhelbredelig kromosomal patologi, der fratager patienter muligheden for at leve et normalt liv. Hvordan et barn med AS vil leve, afhænger i høj grad af typen af kromosomafvigelse.
Duplikering af et kromosomsegment er i de fleste tilfælde uforenelig med liv. Og selvom sådanne patienter ikke dør i spædbarnsalderen og når puberteten, har de ingen chance for at få børn.
Deletion eller fravær af en del af de gener, der forekommer hyppigst ved Angelman syndrom, er en hindring for, at barnet lærer at gå og tale. Sådanne børn har en mere alvorlig form for mental retardering, og epileptiske anfald forekommer oftere, og deres intensitet er meget større end hos patienter med andre kromosomale abnormiteter.
Hvis der kun er en mutation af ét gen, kan barnet med den rette opmærksomhed og tilgang lære det grundlæggende om egenomsorg, kommunikation og interaktion i en gruppe, selvom det stadig vil halte bagud i forhold til sine jævnaldrende i udvikling.
For børn med Angelman syndrom, som er venlige af natur, er det vigtigste forældrenes kærlighed og opmærksomhed. Kun i dette tilfælde vil barnets uddannelse bære frugt, selvom den er lille. Patienter med AS vil naturligvis ikke kunne gå i en almindelig skole. De har brug for særlige klasser, hvor børnene først lærer at koncentrere sig, og derefter gradvist får de grundlæggende skolekundskaber.
Diagnosticering Angelmans syndrom
Angelmans syndrom er en medfødt udviklingspatologi. Men på grund af visse omstændigheder er det ofte umuligt at diagnosticere det i spædbarnsalderen og den tidlige barndom. Dette skyldes manglende specificitet og svage symptomer hos spædbørn og børn under 3 år. Og sygdommens forekomst i vores land er ikke så stor, at læger har lært at genkende den blandt sine ligemænd.
Angelmans syndrom hos spædbørn kan vise sig som nedsat muskeltonus, hvilket manifesterer sig i problemer med at spise (svaghed i suge- og synkerefleksen) og senere vanskeligheder med at lære at gå (sådanne børn begynder at gå meget senere). Disse symptomer er de første tegn på en udviklingsforstyrrelse hos barnet, som meget vel kan være forbundet med en kromosomafvigelse. Kun genetisk analyse kan bekræfte denne antagelse.
Der lægges særlig vægt på børn, hvis forældre har forskellige genomiske eller kromosomale lidelser. Sygdommen manifesterer sig trods alt muligvis ikke i starten, og hvis patologien opdages i tide, er det muligt at opnå betydeligt større succes i læringen ved at begynde at arbejde intensivt med barnet, hvilket bremser sygdommens progression.
Hvis forældrene har forskellige kromosomale abnormiteter, udføres genetisk analyse, selv før barnet er født, da SA er en af de patologier, der kan opdages i embryonalstadiet.
Indsamling af materiale til genetisk forskning kan udføres på to måder:
- invasiv (med en vis procentdel af risiko, da det er nødvendigt at penetrere livmoderen for at tage en prøve af fostervand)
- ikke-invasiv (analyse af barnets DNA fra moderens blod).
Følgende undersøgelser udføres derefter:
- fluorescerende in situ-hybridisering (FISH-metoden) – binding af en DNA-probe mærket med et specielt farvestof til det undersøgte DNA, efterfulgt af undersøgelse under et mikroskop.
- analyse af mutationer i ube3a-genet og imprintede gener,
- DNA-methyleringsanalyse ved hjælp af særlige metoder anvendt i genetik.
Genetiske tests giver ret præcise oplysninger i tilfælde af kromosomafvigelser, hvilket betyder, at kommende forældre ved på forhånd, hvad de skal forberede sig på. Der er dog undtagelser. Hos en bestemt gruppe patienter forbliver testresultaterne normale, når alle symptomer, der indikerer patologi, er til stede. Det vil sige, at patologi kun kan identificeres ved nøje at observere barnet fra den tidlige barndom: hvordan det spiser, hvornår det begyndte at gå og tale, om det bøjer benene, når det går osv.
Ud over FISH-metoden kan man blandt de instrumentelle diagnostiske metoder til Angelmans syndrom udpege tomografi (CT eller MR), som hjælper med at bestemme hjernens tilstand og størrelse, og et elektroencefalogram (EEG), som viser, hvordan individuelle dele af hjernen fungerer.
Læger stiller normalt en endelig diagnose i alderen 3-7 år, når patienten allerede har de fleste symptomer, og dynamikken i sygdommens udvikling er synlig.
Hvilke tests er nødvendige?
Differential diagnose
Angelmans syndrom er en genetisk patologi, der stort set ikke har nogen specifikke manifestationer. De fleste symptomer kan i lige grad indikere både AS og andre genetiske patologier.
Differentialdiagnose af Angelman syndrom udføres med følgende patologier:
- Pitt-Hopkins syndrom (patienter er karakteriseret ved mental retardering, munter karakter, smilende, de har en ret stor og bred mund, mikrocefali er bemærket). Forskellen er anfald af hyperventilation og åndedrætsbesvær i vågen tilstand.
- Christiansons syndrom (patienter er mentalt retarderede personer med en munter disposition, ude af stand til at tale, karakteriseret ved mikrocefali, ataksi, kramper, ufrivillige muskelbevægelser).
- Mowat-Wilsons syndrom (symptomer: mental retardering, epileptiske anfald, spids hage, åben mund, glad ansigtsudtryk, mikrocefali). Kendetegn: stor afstand mellem øjnene, øjnene skråt indad, afrundet næsespids, bagudvendt øremuskel.
- Kabuki-syndrom (karakteriseret ved mild til moderat mental retardering, tale- og motoriske problemer, muskelsvaghed, epileptiske anfald, mikrocefali, lange kløeintervaller og nedsat koordination). Karakteriseret ved buede øjenbryn, udadbøjet lateral del af det nedre øjenlåg, vidtstående øjne, lange øjenhulespalter med lange, tykke øjenvipper.
- Rett syndrom (differentiering fra AS hos kvinder). Symptomer: forsinket taleudvikling, anfald, mikrocefali. Forskellen er, at der ikke er et glad udtryk i ansigtet, der er anfald af apnø og apraksi, som udvikler sig over tid.
- Autosomalt recessivt mentalt tardationssyndrom 38 (symptomer: markant mental retardering med forsinkelser i motoriske færdigheder og tale, muskelsvaghed, spiseproblemer i spædbarnsalderen, impulsivitet). Kendetegnende er iris' blå farve.
- MECP 2-genduplikationssyndrom (differentiering fra SA hos mænd). Symptomer: svær mental retardering, muskelsvaghed siden barndommen, taleproblemer eller mangel på tale, epilepsi. Kendskaber: progressiv myopati, konstant tilbagevendende infektioner.
- Kleefstra syndrom (symptomer: tale- og tankeproblemer, muskelsvaghed, søvnforstyrrelser, manglende opmærksomhed, åben mund, hyperaktivitet, anfald, ataksi, balanceforstyrrelser). Særlige træk: fladt ansigt, kort, stump næse, vidtstående øjne, stor, udadbøjet underlæbe, aggressive udbrud.
- Smith-Magenis syndrom (karakteriseret ved anfald, søvnproblemer, intellektuelle og motoriske udviklingsforstyrrelser). Kendetegnende træk omfatter et bredt og fladt ansigt og en fremtrædende pande.
- Koolen-de Vries syndrom (mild til moderat mental retardering, muskelsvaghed, anfald, venlighed). Kendetegn: langt ansigt med høj pande, udstående ører, skrå øjne, høj ledmobilitet, medfødte hjertefejl.
- Phelan-McDermid syndrom (symptomer: mental retardering, taleforstyrrelser eller mangel på tale). Kendetegn: store hænder med udviklede muskler, muskelsvaghed fra fødslen, svag svedtendens.
Sådanne patologier som adenylsuccinatmangel, autosomalt recessivt mentalt retardationssyndrom 1, kromosom 2q23.1 duplikationssyndrom, FOXG1, STXBP1 eller MEF2C gen haploinsufficienssyndromer og nogle andre kan "prale" af symptomer svarende til Angelman syndrom.
Lægens opgave er at stille en præcis diagnose, differentiere Angelman syndrom fra patologier med lignende symptomer og ordinere effektiv behandling, der er relevant for det diagnosticerede stadium af sygdommen.
Behandling Angelmans syndrom
Angelmans syndrom er en af de patologier, som medicinen stadig søger efter effektiv behandling for. Den ætiologiske behandling af sygdommen er i udviklingsfasen med forskellige metoder og midler, hvoraf mange endnu ikke er blevet testet på mennesker. Det betyder, at lægerne for nuværende er nødt til at begrænse sig til symptomatisk behandling, som på en eller anden måde hjælper med at afhjælpe den ubehagelige situation for børn og voksne med marionetsyndrom, der lider af epileptiske anfald, spytflåd, hypotension og søvnforstyrrelser.
Det er således muligt at reducere hyppigheden og styrken af epileptiske anfald ved hjælp af et korrekt valgt antikonvulsivt lægemiddel. Men hele vanskeligheden er, at anfald hos patienter med SA adskiller sig fra almindelige epileptiske anfald ved, at de er karakteriseret ved flere typer anfald, hvilket betyder, at tilstanden kan lindres ved at administrere flere lægemidler på én gang.
De mest populære antikonvulsiva, der anvendes til behandling af Angelmans syndrom, er: valproinsyre, topiramat, lamotrigin, levetiracetam, clonazepam og lægemidler baseret på disse. Mindre almindeligt anvendte er lægemidler baseret på carmazepin, phenytoin, phenobarbital og ethosuximid, da nogle af dem kan fremkalde en paradoksal effekt, der består i at forstærke og øge hyppigheden af epileptiske anfald. Dette sker, hvis lægemidlet anvendes som en del af monoterapi.
Til behandling af savlen anvendes normalt to metoder: medicinsk (lægemidler, der undertrykker spytproduktionen) og kirurgisk, som involverer reimplantation af spytkanalerne. Men i tilfælde af SA anses disse metoder for at være ineffektive, og spørgsmålet forbliver åbent. Forældre og dem, der passer sådanne patienter, skal være særligt opmærksomme på dette problem, da patienterne normalt ikke selv kontrollerer savlen, og nogle simpelthen ikke er i stand til at tage vare på sig selv.
Et andet problem er kort søvnvarighed. Ofte sover børn med Angelman syndrom ikke mere end 5 timer, hvilket har en negativ indvirkning på hele kroppens funktion. Let ophidsede, aktive børn, der elsker spil og kommunikation (selvom de forsøger at begrænse sig til nonverbale metoder), er mærkbart trætte i løbet af dagen. For at få en god hvile har kroppen brug for en dyb, fuld søvn, men det er netop hage ved det.
Det ser ud til, at beroligende lægemidler (phenothiaziner og atypiske antipsykotika), der beroliger nervesystemet, burde være tilstrækkelige til at forbedre søvnen hos ophidsede patienter. Men i tilfælde af AS er brugen af sådanne lægemidler behæftet med forekomsten af negative virkninger. Derfor foretrækker læger stadig milde sovepiller, såsom melatonin (et naturligt hormonelt lægemiddel baseret på søvnhormonet), som gives til patienter en time før sengetid i en mængde på 1 tablet, og diphenhydramin. Hyppigheden af administration og dosering bestemmes af lægen afhængigt af patientens tilstand og alder.
Nogle gange har patienter med Angelman syndrom problemer med fordøjelsen og afføringen. Du kan forbedre din afføring med afføringsmidler (helst naturlægemidler).
Eller man kan gribe problemet an på en anden måde, som amerikanske læger gjorde, baseret på nogle metoder til behandling af autisme, fordi mange symptomer, der er karakteristiske for AS, også er karakteristiske for autisme (impulsivitet, ufrivillige bevægelser, gentagne handlinger, opmærksomhedsunderskud, kommunikationsproblemer osv.). Det blev bemærket, at introduktionen af hormonet secretin, som normaliserer fordøjelse og afføring, har en positiv effekt på patienternes opmærksomhed, og oxytocin hjælper med at forbedre barnets kognitive evner og hukommelse samt korrekt adfærd.
Sandt nok er hormoner alene ikke nok, især når det kommer til børn. Ved Angelman syndrom er adfærdsterapi, arbejde med en psykolog og talepædagog (undervisning i nonverbale kommunikationsmetoder og tegnsprog) indiceret. Uddannelsen af sådanne børn bør baseres på et individuelt program med deltagelse af specialuddannede lærere, en psykolog og forældre. Desværre er dette ikke muligt alle steder, og familier står alene med deres problem.
Da mange unge patienter med AS lider af lav muskeltonus og ledproblemer, lægges der stor vægt på fysioterapi. Oftest tyr lægerne til brug af paraffinbehandling, elektroforese og magnetisk terapi.
Aktiv tonisk massage og specielle øvelser inden for terapeutisk fysisk træning vil hjælpe det syge barn med at stå på benene og gå selvsikkert efter et stykke tid. Vandgymnastik er især nyttigt i denne henseende, hvilket anbefales til SA i koldt vand. Det øger muskeltonus og lærer barnet at kontrollere sin krop og koordinere bevægelser.
Antikonvulsiv behandling
Det farligste symptom på Angelmans syndrom er anfald, der ligner anfald ved epilepsi. Dette symptom observeres hos 80% af patienterne, hvilket betyder, at de alle skal ordineres effektiv antikonvulsiv behandling.
Behandling af epileptiske anfald udføres ved hjælp af vitaminer og antikonvulsiva. Ved Angelman syndrom, ledsaget af konvulsivt syndrom, vil vitaminer fra gruppe B samt vitamin C, D og E være nyttige. Men det er meget farligt at ordinere vitaminbehandling på egen hånd i dette tilfælde, fordi ukontrolleret indtagelse af vitaminer kan reducere effektiviteten af antiepileptiske lægemidler og fremkalde nye, mere alvorlige og langvarige anfald.
Valget af antikonvulsiv medicin og ordinationen af deres effektive dosis bør også foretages af en speciallæge. Han eller hun afgør også, om ét lægemiddel vil være nok, eller om patienten skal tage 2 eller flere lægemidler i lang tid.
Til de fleste patienter ordinerer læger valproinsyrelægemidler (Valproinsyre, Depakine, Convulex, Valparin osv.), som forebygger anfald og forbedrer patienternes humør og mentale tilstand.
Valproinsyre fås i form af tabletter, sirup og injektionsopløsninger. Det mest populære lægemiddel er depotmedicinen "Depakine" i tabletter og som en opløsning til intravenøs administration. Doseringen af lægemidlet bestemmes individuelt af lægen afhængigt af patientens vægt, alder og tilstand.
Lægemidlet tages under måltider 2 til 3 gange dagligt. Den gennemsnitlige daglige dosis er 20-30 mg pr. 1 kilogram af patientens vægt, den maksimale dosis er 50 mg/kg pr. dag.
Kontraindikationer for brug. Må ikke anvendes i tilfælde af lever- og bugspytkirteldysfunktion, hæmoragisk diatese, hepatitis, porfyri og overfølsomhed over for lægemidlet.
Bivirkninger omfatter håndrystelser, fordøjelses- og afføringsforstyrrelser og ændringer i kropsvægt.
"Topiramat" er også et lægemiddel til behandling af SA. Det produceres i tabletform og anvendes både som en del af monoterapi og i kombination med andre lægemidler.
Administrationsmåde og dosering. Tag tabletterne oralt uanset fødeindtag. Den daglige startdosis for voksne er 25-50 mg, for børn - 0,5-1 mg/kg. Dosis øges hver uge i henhold til lægens anvisninger.
Lægemidlet bør ikke tages under graviditet og amning, samt i tilfælde af overfølsomhed over for dets komponenter. Lægemidlet har mange forskellige bivirkninger.
Lægemidler som en læge kan ordinere mod Angelmans syndrom: Clomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra osv.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditionel medicin og homøopati
Traditionel medicin, ligesom homøopatiske præparater, er naturligvis relativt sikker, men effektiviteten af en sådan behandling af Angelmans syndrom kan betragtes som kontroversiel.
Selvom folkemedicin stadig kan hjælpe med nogle ting, taler vi om at stoppe epileptiske anfald. I den henseende kan urtebehandling være ret effektiv.
En god effekt opnås af en medicinsk samling baseret på pæon, lakrids og andemad (komponenterne tages i lige store mængder). Urterne skal males til mel. Efter 2 uger fra starten af indtagelsen kan man bemærke et betydeligt fald i hyppigheden af anfald.
Lavendelafkog (1 tsk pr. glas kogende vand) er også nyttigt mod kramper. Blandingen koges i 5 minutter og trækkes i en halv time. Medicinen tages om natten i 14 dage.
En vandig (eller alkoholisk) infusion af motherwort anses for effektiv til epileptiske anfald.
Af de homøopatiske præparater til forebyggelse af anfald ved Angelman syndrom kan man bruge lægemidler baseret på kamille og motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Men det skal tages i betragtning, at kun en homøopatisk læge kan ordinere effektive og sikre doser af lægemidler i hvert enkelt tilfælde.
Forebyggelse
Som læseren sikkert allerede har forstået, er medicinen endnu ikke i stand til at forhindre genmutationer og andre kromosomafvigelser, samt at korrigere situationen. Dette kan ske for alle, fordi børn med Angelman syndrom fødes af raske forældre, og genetik, som i øjeblikket er en af de mindst studerede grene af medicinen, kan endnu ikke forklare dette.
Det eneste, der kan gøres, er at tage en ansvarlig tilgang til graviditetsplanlægning, registrere sig og gennemgå undersøgelser i tide. Men igen, en sådan foranstaltning vil være mere pædagogisk end forebyggende, ligesom enhver anden undersøgelse. Men unge forældre vil vide på forhånd, hvad de skal forberede sig på, og i tilfælde af et positivt svar vil de beslutte, om de kan påtage sig et sådant ansvar som at opdrage et sygt barn.
Vejrudsigt
Prognosen for Angelman syndrom afhænger af kromosomafvigelsens art og hvor hurtigt den opdages. De hårdest ramte er de børn, hvis kromosom 15 indeholder "huller" i gener (deletion). Sandsynligheden for, at sådanne patienter kan gå og tale, er ekstremt lav. Andre tilfælde kan korrigeres med en omhyggelig tilgang og kærlighed til dit barn.
Desværre vil sådanne patienter ikke være i stand til at blive fuldgyldige medlemmer af samfundet, på trods af at de langt fra er dumme, de forstår tale og dens betydning. De vil dog have problemer med kommunikation resten af deres liv. Patienter kan lære tegnsprog fra barnsben, men de kan ikke tvinges til at kommunikere ved hjælp af ord. Ordforrådet hos "talende" patienter er begrænset til et minimum af ord, der bruges i hverdagen (5-15 ord).
Hvad angår forventet levetid og generel sundhed for patienter med Angelman syndrom, svinger tallene her omkring gennemsnitsværdier. I voksenalderen står patienterne oftest over for helbredsproblemer som skoliose og fedme, som med den rette behandlingstilgang ikke er livstruende.