Medicinsk ekspert af artiklen

Nye publikationer
Amaurotisk idioti
Last reviewed: 04.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.

Amaurotisk idioti er en sjælden, progressiv sygdom. Den er karakteriseret ved en gradvis nedgang i synet til fuldstændig blindhed og forringelse af intelligensen, indtil idiotien indtræder. Som følge heraf udvikler patienten dyb marasmus med dødelig udgang. Sygdommen blev først beskrevet af øjenlægen Dr. Tau for mere end 130 år siden. Tau bemærkede en særlig transformation af fundus. Mere end 500 tilfælde af sygdommen er allerede blevet beskrevet.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Epidemiologi
Årsager Amaurotisk idioti
Trods de mange indsamlede data om sygdommen fortsætter forskere i øjeblikket med at søge efter svar på mange spørgsmål om årsagerne, patogenesen og endda manifestationerne af amaurotisk idioti.
Der er forslag om, at sygdommen er arvelig. Arvemåden er autosomal recessiv. Oftest påvirkes lillehjernen og nakkelapperne i hjernehalvdelene, med alvorlige konsekvenser og komplikationer for hele kroppen: atrofi af synsnerverne, nervefibre kan miste deres membraner, og forbindelser mellem nerveceller kan gå i opløsning.
De fleste eksperter anerkender, at de kliniske tegn på sygdommen kan være ret varierede og korrelere med den alder, hvor amaurotisk idioti begyndte at udvikle sig hos patienten.
Under undersøgelsen af årsagerne til sygdommen blev der bemærket et bestemt mønster: sygdommen rammer ofte børn fra samme familie, hvilket er grunden til, at navnet "familiær amaurotisk idioti" bruges. Ifølge undersøgelser, hvis resultater blev offentliggjort, da man lige var begyndt at studere sygdommen, blev der ud af 64 tilfælde af amaurotisk idioti fundet 37 i 13 familier (hver familie havde 2-5 syge børn). Det er bemærkelsesværdigt, at de syge i sådanne familier havde absolut raske brødre og søstre. I dag menes det, at faktoren recessiv arv spiller en stor rolle i sygdommens udvikling. Det er således muligt at forklare hyppigheden af forekomsten af tilfælde af sygdommen i de samme familier. Når man analyserer arvelighedsfaktoren som årsag til amaurotisk idioti, må man ikke begrænse sig til tilstedeværelsen af klinisk udtrykte tegn i patientfamilier (både i de ascenderende og laterale linjer), men også tage hensyn til rudimentære, for eksempel karakteristiske afvigelser i det visuelle apparats funktion (familiær choroiditis, pigmenteret retinal dystrofi osv.).
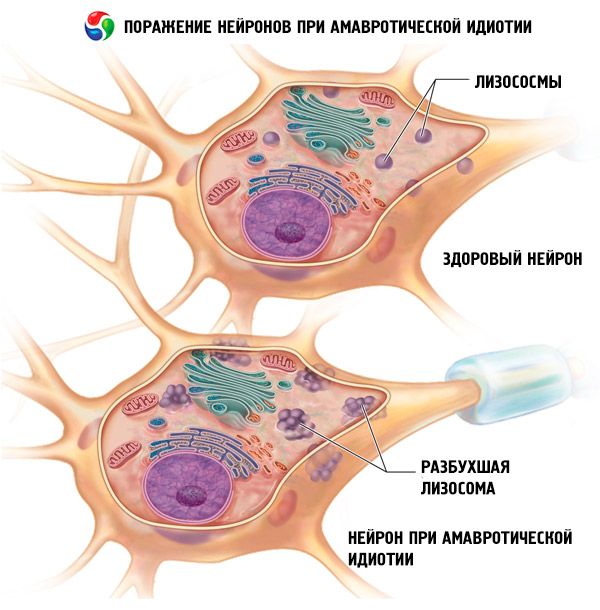
Symptomer Amaurotisk idioti
Niveauer
Den infantile form udvikler sig fra 4-6 måneder. Denne form for amaurotiske idiopatier er karakteriseret ved en familiær natur. Synet forringes hurtigt: babyen kan ikke fiksere blikket, observerer ikke objekter. Den såkaldte "kirsebærgrube" vises på fundus - en rødlig plet i makulaområdet, som er omgivet af en gråhvid kant. Derefter atrofierer synsnerverne, og barnet mister fuldstændigt evnen til at se. Orientering, beskyttelsesreflekser samt evnen til at bevæge sig går gradvist tabt. Patienterne reagerer kraftigt på lydstimuli - de krymper sig ved en lyd, der er stille for en rask person, kramper kan observeres på grund af øget muskeltonus. I sygdommens sidste fase udvikles generel atrofi, udmattelse af kroppen og øget tonus i alle ekstensormuskler. Prognosen for sygdommen er også skuffende: patienten dør halvandet til to år efter sygdommens debut.
Den sene barndomsform begynder i 3-4-årsalderen. Den progressive sygdom veksler med stadier af remission. Det gradvise tab af intelligens ledsages af anfald, koordinationsforstyrrelser og ekstrapyramidale lidelser. Denne form er også karakteriseret ved atrofi af synsnerven. Døden indtræffer 6-8 år efter starten af amaurotisk idioti.
Den juvenile form begynder at manifestere sig i 6-10-årsalderen. Spielmeyers amaurotiske idioti udvikler sig langsommere. Ændringer i fundus falder sammen med manifestationer af pigmentær retinal dystrofi. Patientens syn falder gradvist, ligesom intelligensen. Nedsatte motoriske funktioner kan manifestere sig på forskellige måder og uregelmæssigt: ikke særlig udtalt lammelse af arme og ben, ekstrapyramidale og bulbære lidelser forekommer. Sygdommen fører til døden 10-25 år efter udviklingen af de første tegn.
Den sene form forekommer meget sjældent og udvikler sig ekstremt langsomt. Patientens mentale tilstand ændrer sig (som et organisk mentalt syndrom), atrofi af synsnerverne og pigmentdystrofi af nethinden observeres. Det sidste stadie er karakteriseret ved lammelse og epileptiformt syndrom. Patienten dør 10-15 år efter sygdommens debut.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Forms
Der er fire typer amaurotisk idioti:
- Tay-Sachs (påvirker i en tidlig alder);
- Jansky-Bilynovsky (forekommer hos børn i en senere alder);
- Spielmeyer-Vogt syndrom (forekommer hos unge);
- Kufsa (sen form).
Nogle forskere skelner også den medfødte Norman-Wood-type separat.
Hver type sygdom har sit eget sæt af kliniske manifestationer, men de er alle forenet af fælles årsager, klinisk billede, anatomisk base og patogenese.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnosticering Amaurotisk idioti
Amaurotisk idioti skyldes en forstyrrelse i lipidmetabolismen, hvorved et mellemprodukt af lipidmetabolismen, sphingomyelin, aflejres i forskellige celler i kroppen. Placeringen og sammensætningen af aflejringerne bestemmer udviklingen af et specifikt klinisk billede af sygdommen.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Hvordan man undersøger?
Differential diagnose
Differentialdiagnose af amaurotisk idioti er baseret på et specifikt klinisk billede og karakteristiske patologier i fundus.
Den tidlige form har symptomer, der ligner Landings sygdom, en type mukopolysakkaridose. Landings sygdom udvikler sig fra de første måneder efter fødslen og fører til døden efter 2-3 år. En "kirsebærkerne" på fundus optræder i 1/5 af tilfældene, degenerative forandringer i nethinden og forvrænget opfattelse af lyde (hyperkussion) er praktisk talt ikke karakteristiske for den, men samtidig forstørrelse af milt og lever, psykiske lidelser og bevægelsesforstyrrelser observeres.
Den juvenile form overlapper undertiden med manifestationerne af Lawrence-Moon-Biedl syndrom. For at differentiere disse sygdomme er det nødvendigt at være opmærksom på deres andre manifestationer. Lawrence-Moon-Biedl syndrom er karakteriseret ved hurtig vægtøgning, deformation af lemmer, der er karakteriseret ved tilstedeværelsen af yderligere fingre eller tæer, mærkbare vegetative-trofiske lidelser og fravær af motoriske funktionsforstyrrelser.
De mange forskellige symptomer på sen amaurotisk idioti komplicerer diagnosen i løbet af livet. Dens manifestationer ligner Friedreichs ataksi, multipel sklerose, Alzheimers sygdom, Picks sygdom, progressiv lammelse og endda skizofreni.
Nogle forfattere insisterer på, at diagnosen af denne sygdom, især når de kliniske manifestationer er slørede, kun kan stilles pålideligt efter døden, baseret på analyse af histologiske abnormiteter i nervesystemet.
Hvem skal kontakte?
Behandling Amaurotisk idioti
Der findes ingen rationel og effektiv behandling. I dag er terapi for amaurotisk idioti udelukkende rettet mod at lindre symptomer. Der anvendes beroligende midler, nootropika, antikonvulsiva og generelle tonika.
For at aktivere blodcirkulationen og metaboliske processer i hjernen ordineres glycin, elkar, cerebrolysin, glutaminsyre og pantogam.
For at lindre konvulsivt syndrom ordineres difenin eller karmazepin.
Et positivt resultat kan opnås ved brug af vævsekstrakter, blodtransfusion eller plasma.
Forebyggelse
Manglen på effektiv behandling af amaurotisk idioti tvinger os til at være meget opmærksomme på forebyggelse. Der findes allerede metoder, der giver os mulighed for at identificere heterozygote bærere af det patologiske gen, og metoder til diagnosticering af amaurotisk idioti under graviditet. Prænatal diagnostik af sygdommen består i at analysere aktiviteten af hexosaminidase A i fostervandet. Hvis der opdages reduceret enzymaktivitet, anbefales det at afbryde graviditeten. Forældre til et sygt barn rådes til at stoppe med at få børn.
Использованная литература