Medicinsk ekspert af artiklen

Nye publikationer
Spinal muskelatrofi
Sidst revideret: 07.06.2024

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Spinal muskelatrofi er ikke en enkelt nosologisk enhed, men en hel gruppe af klinisk og genetisk heterogene arvelige patologier fremkaldt af de stigende processer med degeneration af motoneuroner i de forreste spinalhorn. Udtrykket omfatter forskellige varianter af genetisk bestemt perifer parese og muskelatrofi som følge af degeneration af spinale motoriske neuroner og/eller hjernestamme. Den mest almindelige årsag til problemet er en autosomal recessiv mutation på den lange q-skulder af det femte kromosom. Behandlingen er uspecifik, rettet mod at forbedre troficiteten af nervevævet og yde palliativ støtte for at forbedre livskvaliteten.[1]
Epidemiologi
Spinal muskelatrofi forekommer i ét tilfælde pr. 6.000 til 10.000 nyfødte (ifølge American Journal of Medical Genetics 2002).
Prævalensen af SMN-gen exon 7 deletionsbærere er 1:50 personer.
Bulbo-spinal muskelatrofi (Kennedy syndrom) forekommer hos et barn ud af 50.000 og er den mest almindelige voksne type spinal amyotrofi.
Det bemærkes, at halvdelen af børnene med denne sygdom ikke overvinder den toårige overlevelsesperiode.
Patologien nedarves efter det autosomale recessive princip. Oftest er hver forælder til et sygt barn bærer af en kopi af det muterede gen. Da mutationen kompenseres for ved tilstedeværelsen af en anden "normal" genkopi, har forældrene ingen manifestationer af spinal muskelatrofi. Type 2-patologi arver normalt ikke en ekstra kopi fra forælderen. Problemet opstår på grund af en utilsigtet fejl under dannelsen af kønsceller eller direkte på tidspunktet for befrugtning. Med spinal muskelatrofi af den første type forekommer spontan udvikling af sygdommen kun i 2% af tilfældene (i denne situation er bæreren kun en af forældrene).[2]
Årsager Spinal muskelatrofi
Hovedårsagen til spinal muskelatrofi er en mutation af genet, der er ansvarlig for produktionen af SMN-protein lokaliseret på kromosom 5q. Denne lidelse forårsager yderligere den gradvise død af motoriske nerveceller i de forreste horn i rygmarven og hjernestammen. Som et resultat af disse processer falder tonen i muskulaturen, atrofi af respiratoriske, svælg-, ansigts- og skeletmuskler udvikler sig. Den dominerende type arv af pædiatriske former for spinal muskelatrofi er autosomal recessiv, hvilket indebærer, at begge forældre bærer defekte gener samtidig. Hvad angår type IV-patologi (voksen form), er der en forbindelse til X-kromosomet, så kun mænd er påvirket.
Udviklingen af spinal muskelatrofi er baseret på de stigende processer med degeneration og død af motoriske neuroner i de spinale forhorn, beskadigelse af hjernestammens kerner. Patologiske ændringer er mest intense i zonerne med cervikal og lumbal fortykkelse. Det cellulære antal reduceres til et minimum, udskiftning af bindevæv sker, hvilket skyldes svigt af celledødsprogrammet - den såkaldte apoptose. Ændringen påvirker strukturerne af de motoriske kerner af kranienerver, forreste rødder, motoriske nerver. Der er en klinik for neurogen fascikulær atrofi. Med et forlænget sygdomsforløb på et sent stadium af bindevævsovervækst opstår.
Udseendet af det tilsvarende kliniske billede er forbundet med en mangel på SMN-proteinet, som påvirker den vellykkede funktion af motoriske nerveceller i de forreste spinalhorn. Proteinmangel som et af led i udviklingen af spinal muskelatrofi blev opdaget i slutningen af det XX århundrede. På baggrund af motoneuronskade er innervation af skeletmuskler (hovedsagelig proksimale sektioner) svækket.[3]
Risikofaktorer
Mangfoldigheden af kliniske former for spinal muskelatrofi 5q forklares ved tilstedeværelsen af visse modificerende faktorer, der kan opdeles i to kategorier: dem, der påvirker og dem, der ikke påvirker SMN-proteinscore.
- I øjeblikket anses SMN2-genet for at være den grundlæggende faktor i udviklingen af spinal muskelatrofi: Jo flere kopier af SMN2-genet, jo lavere er intensiteten af sygdomssymptomerne. Den anden faktor, som er direkte relateret til den centromere kopi af SMN-genet, er en 1-nukleotidsubstitution c.859G>C i exon 7 af SMN2-genet, hvilket fører til dannelsen af et nyt enhancer-bindende splejsningssted: resultatet er inklusion af exon 7 i transkriptet fra SMN2-genet. Denne variation er forbundet med en stigning i blodniveauet af fuldlængde SMN-protein hos patienter med spinal amyotrofi af anden eller tredje type.
Andre faktorer, der påvirker antallet af SMN'er:
- Splejsningsregulerende faktorer (Tra2β - inducerer exon-springning af exon 7, SF2/ASF - øger exon 7-inklusion, hnRNPA1 - undertrykker exon 7-inklusion af SMN2-genet).
- Transskriptionsregulerende faktorer (CREB1 - øger SMN-transkription, STAT3 - favoriserer axonvækst, IRF1 - øger SMN-tal, PRL - øger levetiden i alvorlige stadier).
- MRNA-stabiliserende faktorer (U1A -reducerer SMN, HuR/p38).
- Faktorer, der påvirker post-translationel modifikation (RCA - undertrykker SMN-nedbrydning, GSK3 - øger overlevelse).
- Eksogene faktorer (sult, hypoxi, oxidativt stress).
Virkningerne af de ovennævnte faktorer blev overvejende bestemt in vitro.
- Faktorer, der ikke er forbundet med SMN-genet - især proteiner, der optimerer endocytose ved synapser (laminin 3, coronin, neurocalcin delta, calcium-neurin-lignende protein).
Yderligere opmærksomhed rettes mod DNA-methylering, den mest stabile modifikation, der påvirker arten af genekspression. Methyleringen af en gruppe gener, der muligvis er involveret i patogenetiske processer, viste sig at være korreleret med sværhedsgraden af spinal muskelatrofi.[4]
Patogenese
Spinal muskelatrofi er en genetisk patologi, for hvilken enhver af arvetyperne - både autosomal dominant og autosomal recessiv eller X-bundet - er iboende. Oftest taler vi om autosomal recessiv patologi i tidlig barndom. Ansvaret for dannelsen af sådan spinal amyotrofi er SMN-genet, lokaliseret i locus 5q13. Deletionen af exon 7 i SMN-genet resulterer i patologi med mulig involvering af de nærliggende gener p44 og NAIP.
SNM-genomet koder for et protein, der inkluderer 294 aminosyrer og har en MM på ~38 kDa. Proteinet har følgende funktioner:
- er en del af RNA-proteinkomplekset;
- deltager i dannelsen af spliceosomstedet, der katalyserer præ-RNA-splejsning;
- Involveret i processer, der kontrollerer proteinproduktion og proteinisoformer;
- tilvejebringer aksonal transport af mRNA;
- Begunstiger nervecellevækst og giver neuromuskulær kommunikation.
Et par typer SMN-gener er kendt:
- telomerisk SMNt (SMN1);
- centromerisk SMNc (SMN2).
Langt de fleste tilfælde af spinal muskelatrofi skyldes ændringer i SMN1-genet.
Kennedy spinal muskelatrofi har en kobling til Xq12-locuset, der indeholder NR3C3-genet, som koder for et androgenreceptorprotein. Den har en X-bundet arvevariant. Når antallet af CAG-gentagelser i en gen-exon stiger, udvikler patologien sig.
Undertrykkelse af SNM-proteinproduktion er ledsaget af følgende ændringer:
- på grund af nedsat axonkoordination forekommer overdreven forgrening af axoner;
- væksten af axoner bremses og deres størrelse falder;
- der er ukorrekt klyngning af calciumkanaler i vækstkeglen;
- Uregelmæssige presympatiske terminaler af motoriske nervecelle-axoner dannes.
Rygmarven begynder aktivt at miste motoriske neuroner i de forreste horn, hvilket tegner sig for udviklingen af atrofi af de proksimale lemmermuskler.[5]
Symptomer Spinal muskelatrofi
Symptomatologi af spinal muskelatrofi Werdnig-Hoffman debuterer oftest i perioden med nyfødte og op til seks måneder, manifesteret af syndromet af en "træg" baby. Det klokkeformede bryst, intens hypotoni, mangel på reflekser, muskeltrækninger i tungen og åndedrætsbesvær bemærkes. Syge spædbørn dør oftere, før de når en alder af to år: Dødeligt udfald skyldes stigende respirationssvigt på baggrund af overholdelse af infektiøse processer.
Den mellemliggende form for spinal muskelatrofi af den anden type detekteres fra seks måneders alder. Ud over syndromet af et "trægt" barn er der lavt blodtryk, manglende reflekser, åndedrætsforstyrrelser og tungetrækninger. Selvom børn er i stand til at sidde op, udvikles der flere kontrakturer i de store led.
Kugelberg-Wielander spinal muskelatrofi starter også i den tidlige barndom, med børn i stand til at bevæge sig selvstændigt. Der er svækkelse af iliaca, quadriceps og adduktormuskler, lavt blodtryk, nedsatte reflekser og tungetrækninger. Mange patienter mister evnen til at bevæge sig (gå) selvstændigt med årene.
Spinal muskelatrofi type 4 starter i en ældre alder. Det er karakteriseret ved langsom progression og relativt godartet prognose.[6]
Kennedy-atrofi manifesterer sig oftest i middelalderen (kan generelt debutere hos patienter i alderen 15-60 år). Symptomatologi omfatter muskelømhed og muskelsvaghed, gynækomasti, distal svaghed, sløvhed, tungetrækninger og atrofi. Tegn på bulbar dysfunktion er til stede:
- synkebesvær;
- forhåbning;
- svækkelse af tyggemusklerne;
- dysartri;
- posturale og motoriske rystelser i hænderne.
Første tegn på androgenmangel:
- gynækomasti (hos omkring 60% af patienterne), ofte asymmetrisk;
- forringelse af seksuel funktion (oligospermi, testikelatrofi, erektil dysfunktion).
Første tegn
Spinal amyotrofi manifesteres ved svaghed i muskler og generel impotens. Alle sensoriske og intellektuelle evner påvirkes ikke.
Vigtigste indeks for neuromuskulær patologi:
- muskulatur "doven", svækket, slaphed og slaphed af muskler er noteret;
- muskeltonus er lav, senereflekser er minimeret eller fraværende;
- normale eller fraværende plantarreflekser;
- Korte trækninger af individuelle muskelgrupper noteres (kan ses under huden, på tungen);
- der er tegn på muskelatrofi.
Werdnig-Hoffmans syndrom manifesteres af udtalt hypotoni af musklerne, generel sløvhed, barnets manglende evne til at holde hovedet, vende om og sidde op. Når man forsøger at støtte barnet i maveområdet i en suspenderet tilstand, ser kroppen ud til at "sænke". Hoste-, synke- og sutterefleks er utilfredsstillende, mad kommer ofte ind i luftvejene, vejrtrækning er problematisk. Der kan være ledforvridning forbundet med intrauterin hypotoni. Anamnestiske oplysninger indsamlet under graviditeten indikerer ofte lav fosteraktivitet.
Grundlæggende tegn på spinal muskelatrofi type I:
- alvorlig retardering i motorisk udvikling;
- Hurtig indtræden af ledkontrakturer og thoraxkrumning;
- stigende luftvejs- og bulbar lidelser, problemer med at synke (både mad og spyt) og ekspektorering af sputum;
- øget risiko for aspirationsbetændelse;
- infektion, progressiv respirationssvigt.
Spinal muskelatrofi type II manifesteres af en klar hæmning af motorisk udvikling. Selvom mange patienter kan sidde uden hjælp, og nogle gange endda kravle og stå, går disse evner ofte tabt over tid. Fingerrystelser, muskel- og ledforvridninger (knogle) og åndedrætsproblemer er noteret. Mulig kalvepseudohypertrofi.
Hovedtræk ved type II patologi:
- udviklingsforsinkelser, herunder at standse og vende udviklingen af allerede erhvervede færdigheder og evner;
- øget svaghed i de interkostale muskler;
- overfladiskhed af diaphragmatisk vejrtrækning, svækket hosterefleks, gradvis forværring af respirationssvigt;
- krumning af thorax og rygsøjlen, kontrakturer.
Ved Kugelberg-Wielander syndrom er manifestationerne mildere, langsomt fremadskridende. Patienten er i stand til at bevæge sig rundt, men der er problemer med at jogge eller gå på trapper. Forsinkede symptomer omfatter ofte synke- og tyggebesvær.
Spinal muskelatrofi type IV viser sig allerede i ældre (voksen) alder og er karakteriseret ved det mest "milde" og gunstige forløb. De vigtigste tegn: gradvist tab af evnen til at bevæge sig.[7]
Forms
Spinal muskelatrofi er en del af en gruppe af arvelige patologier karakteriseret ved degenerative ændringer, død af motoriske nerveceller i de forreste spinalhorn og ofte de motoriske kerner i hjernestammen. Processen kan give sig til kende i forskellige livsperioder, det kliniske billede er ikke altid det samme. Arvetyper og forløb kan også være forskellige.
Pædiatrisk spinal muskelatrofi blev først beskrevet så tidligt som i slutningen af det 19. århundrede. Omkring midten af det 20. århundrede blev de vigtigste former for sygdommen identificeret:
- Medfødt (manifesterer sig næsten umiddelbart efter spædbarnets fødsel);
- Tidlig infantil form (opstår på baggrund af tidligere normal udvikling af babyen);
- sen infantil form (afslører sig selv fra en alder af 2 og ældre).
Nogle specialister kombinerer den anden og tredje form til en pædiatrisk type spinal amyotrofi.
Det er generelt accepteret at opdele patologi i pædiatrisk og voksen. Spinal muskelatrofi hos børn klassificeres i tidlig (med en debut i de første par måneder efter barnets fødsel), sen og teenager (ung eller ung). De oftest involverede syndromer er:
- Werdnig-Hoffman atrofi;
- Kugelberg-Wielander-formen;
- kronisk infantil spinal muskelatrofi;
- Vialetto-van Lares syndrom (bulbospinal type med manglende hørelse);
- Fazio-Londe syndrom.
Voksen spinal muskelatrofi debuterer over en alder af 16 år og indtil cirka 60 år, kendetegnet ved en relativt godartet klinik og prognose. Voksenpatologier omfatter:
- Kennedys bulbospinal atrofi;
- scapuloperoneal atrofi;
- facial-lap-skulder og oculo-pharyngeale former;
- distal spinal atrofi;
- monomelisk spinal atrofi.
Separat isoleret og kombineret spinal atrofi. Isoleret patologi er karakteriseret ved overvægt af skader på spinale motoriske neuroner (som ofte er det eneste tegn på problemet). Kombineret patologi er sjælden og repræsenterer et kompleks af neurologiske og somatiske lidelser. Der er beskrivelser af tilfælde af kombineret syndrom med medfødte koronare misdannelser, manglende auditiv funktion, oligofreni, cerebellar hypoplasi.
Spinal muskelatrofi hos ældre er oftest repræsenteret ved Kennedy bulbospinal amyotrofi. Denne patologi er nedarvet recessivt X-bundet. Sygdommens forløb er langsomt, relativt godartet. Det begynder med atrofi af den proksimale muskulatur i underekstremiteterne. Mulig rysten i hænder, hoved. Samtidig opdages også endokrine problemer: testikelatrofi, gynækomasti, diabetes mellitus. På trods af dette forløber patologien hos voksne i en mildere form end hos børn.
En variant af spinal muskelatrofi. |
Patologiens debut |
Detekterbart problem |
Dødsalderen |
Karakteristisk symptomatologi |
Spinal muskelatrofi type 1 (andet navn Verding-Hoffman spinal muskelatrofi) |
Fra fødslen til seks måneder |
Barnet kan ikke sidde op |
Op til to år |
Alvorlig muskelsvaghed, hypotoni, problemer med at holde hovedet op, svækket gråd og hoste, synke- og spytproblemer, udvikling af respirationssvigt og aspirationspneumoni |
Spinal muskelatrofi type 2 |
Seks måneder til halvandet år |
Babyen kan ikke holde ud |
Mere end to år |
Motorisk retardering, vægtmangel, hostesvaghed, håndrystelser, spinal krumning, kontrakturer |
Spinal muskelatrofi type 3 (andet navn Kugelberg-Welander spinal muskelatrofi) |
Efter halvandet år. |
Kan i starten stå og gå, men i en vis alder kan denne evne være tabt |
I voksenlivet. |
Svækkede muskler, kontrakturer, hypermobilitet i leddene |
Spinal muskelatrofi type 4. |
Ungdom eller voksenliv |
Kan i starten stå og gå, men i en vis alder kan denne evne være tabt |
I voksenlivet. |
Tiltagende proksimal muskelsvaghed, nedsatte senereflekser, muskeltrækninger (fascikulationer) |
Om distal spinal atrofi siges i tilfælde af læsioner af motoriske nerveceller i rygmarven, som innerverer den nederste del af kroppen. Karakteristiske tegn på sådan patologi er:
- atrofi af lårmusklerne;
- svaghed i knæ, fodstrækkere og hofteadduktormuskler.
Ingen ændring i senereflekser.
Distal spinal muskelatrofi er repræsenteret af to allelvariationer med en overlappende fænotype:
- scapulo-perineal spinal muskelatrofi;
- Arvelig motor-sensorisk neuropati af Charcot-Marie-Tooth type 2C.
Proksimal spinal muskelatrofi 5q er karakteriseret ved stigende symptomatologi af slap lammelse og muskelatrofi, som skyldes degenerative ændringer i de alfamotoriske neuroner i de forreste spinalhorn. Medfødt sygdom med postpartum asfyksi er den mest alvorlige form: fra det øjeblik, barnet er født, er motorisk aktivitet praktisk talt fraværende, der er kontrakturer, synke- og åndedrætsproblemer. I de fleste tilfælde dør et sådant barn.[8]
Komplikationer og konsekvenser
Yderligere progression af spinal amyotrofi fører til svaghed og reduktion af muskelmassen i lemmerne (især benene). Babyen har i starten ikke eller mister gradvist erhvervede færdigheder - det vil sige, mister evnen til at gå, sidde uden støtte. Motorisk aktivitet af de øvre lemmer falder, leddene bliver stive, over tid er kontrakturer knyttet, og rygsøjlen bliver buet.
For at bevare motoriske evner så længe som muligt og forhindre udvikling af komplikationer anbefales det:
- øv korrekt kropsholdning (anti-tyngdekraftsposition), både i sengen og når du sidder, går osv..;
- regelmæssig fysioterapi, strækøvelser, massage, fysioterapi, uanset typen af spinal muskelatrofi;
- brug specielle senge, stole (kørestole), madrasser og puder;
- Vælg og brug støttende orthotics, korsetter;
- udøve hydroterapi og kinesioterapi, som har en gunstig effekt på åndedræts-, muskuloskeletale- og fordøjelsesapparatet, nerve- og kardiovaskulære system;
- Udfør regelmæssige diagnostiske kontroller, herunder kliniske tests, ryg- og bækkenrøntgenbilleder;
- systematisk konsultere en fysioterapeut og ortopæd med erfaring i at arbejde med lignende patienter;
- Juster korsetter, ortoser, ortopædiske apparater, kørestole osv. Afhængigt af dynamikken.
Pårørende til en patient med spinal muskelatrofi bør gøres bekendt med:
- med det grundlæggende i sikker adfærd, fysioterapi, massage, fysioterapi;
- med reglerne for at opretholde patientens uafhængige aktivitet, brug af ortopædiske anordninger;
- med reglerne for pleje, hygiejne.
Spinal amyotrofi er ofte kompliceret af nedsat tygning, synkning og ledning af mad, som truer aspiration og udvikling af aspirationsbetændelse i lungerne eller obstruktion af luftvejene, som er mest karakteristisk for patologien af den første type. Synkeproblemer ses af symptomer som signifikant og vedvarende forlængelse af spiseperioden, modvilje mod at spise, mad, der falder ud af munden, regelmæssig gagging og forværret vægttab.
Forstyrrelser af fordøjelsesmotilitet afslører sig selv forstoppelse, svag peristaltik, længerevarende ophold af mad i maven (gastrisk stase), udvikling af gastroøsofageal refluks. For at forhindre sådanne komplikationer er det nødvendigt:
- overvåg patientens korrekte position, mens du spiser;
- Brug eventuelt en gastrisk sonde eller gastrostomi for at sikre tilstrækkelig væske- og næringsstofindtagelse og reducere risikoen for aspiration;
- overhold reglerne for tilberedning af mad og drikke, hold øje med deres konsistens og hyppigheden af måltider;
- afhængig af lægens ordination, brug medicin, massage, fysioterapi mv.
En af de mest alvorlige komplikationer af spinal amyotrofi er respiratorisk dysfunktion forbundet med svaghed i åndedrætsmusklerne. Luftvejslidelser kan være dødelige, både hos spædbørn med type 1 patologi og hos unge og voksne patienter med type 2 eller 3 sygdom. De vigtigste problemer er som følger:
- hosterefleks er forstyrret, der er problemer med ekspektorering af sputum fra luftvejene;
- Stigende underskud i mængden af luft, der kommer ind i lungerne, nedsat udskillelse af kuldioxid fra lungerne;
- forvrænger brystet, komprimerer og deformerer lungerne;
- infektiøse processer i form af bronkopneumoni.
For at forhindre sådanne komplikationer anbefales patienter ofte at udføre åndedrætsøvelser ved hjælp af en Ambu-taske.[9]
Diagnosticering Spinal muskelatrofi
Hos patienter med mistanke om spinal amyotrofi er undersøgelser som disse af diagnostisk værdi:
- blodkemi;
- genetisk DNA-analyse;
- elektroneuromyografi.
Blandt yderligere metoder er det muligt at udpege en biopsi af muskelfibre, ultralyd og resonansbilleddannelse af muskulaturen og hjernen.
Blodprøver kan indikere, at kreatinfosfokinase er fysiologisk normalt, men i nogle tilfælde kan det være forhøjet til omkring 2,5 gange.
Elektroneuromyogrammet afslører ændringer som følge af tab af motoriske spinale neuroner. Dette detekteres ved et fald i interferenskurvens amplitude, forekomsten af spontane aktive potentialer, som er fibrilleringer og fasciokulationer, der danner en specifik "frekvensrytme". Hastigheden af impulssignalet, der passerer gennem perifere motorfibre, er normal eller nedsat på grund af sekundære denerveringsforstyrrelser.[10]
Instrumentel diagnose er ofte også repræsenteret ved ultralyd eller MR af muskulaturen, som muliggør påvisning af muskeludskiftning med fedtvæv. MR afslører et typisk patologisk procesmønster, der er unikt for spinal muskelatrofi. Dette er dog kun muligt i de sene stadier af læsionen.
I løbet af morfologisk analyse af muskelbiopsi hos patienter bestemmes et uspecifikt billede i form af bundteatrofi og gruppering af muskelfibre. Det overvældende antal af påvirkede muskelfibre hører til type 1, immunhistologiske og kemiske egenskaber er inden for normale grænser. Det ultrastrukturelle billede er uspecifikt.
Den vigtigste diagnostiske procedure for mistanke om spinal muskelatrofi er test, der kan påvise SMN-genmutationen. Ved direkte DNA-analyse er det muligt at påvise tilstedeværelsen eller fraværet af den syvende og ottende exon af SMNc- og SMNt-generne. Den mest informative metode er kvantitativ analyse, som kan bestemme genkopiantallet og belyse formen af spinal muskelatrofi. Den kvantitative metode er også vigtig i vurderingen af patientens status. Det er en nødvendig foranstaltning, der udføres med henblik på yderligere medicinsk og genetisk familierådgivning.
Yderligere diagnostiske test udføres kun efter et negativt resultat af SMN-gendeletion er opnået. Hvis detektion af punktmutationer er påkrævet, kan direkte automatiseret sekventering af SMNt-genet anvendes.[11]
Differential diagnose
Differentialdiagnosen udføres med patologiske processer, der afslører symptomkomplekset af "træg patient", med medfødte muskeldystrofier, strukturel eller mitokondriel myopati. Især bør tilstedeværelsen af sådanne patologier udelukkes:
- motor neuron sygdom;
- primær lateral myosklerose;
- muskeldystrofi;
- medfødte myopatier;
- sygdomme forbundet med glykogenakkumulering;
- polio;
- autoimmun myasthenia gravis.
Den diagnostiske algoritme udvikles afhængigt af de særlige kendetegn ved symptomatologi hos et bestemt barn. Der anvendes således en særlig klassificering af patienter afhængig af funktionsstatus (Europrotocol TREAT-NMD):
- Kan ikke sidde op uden støtte (sengeliggende).
- Kan sidde, men ude af stand til at gå (siddende).
- Kan bevæge sig selvstændigt (gående patienter).
Følgende diagnostiske algoritme anbefales til patienter i den første gruppe:
- Fysisk undersøgelse (påvisning af brystkrumning, vurdering af åndedræts- og hostefunktion og hudtilstand);
- hjerte- og respirationsovervågning, polysomnografi og identifikation af symptomer på pulmonal ventilationsmangel;
- pulsoximetri for at bestemme graden af iltning;
- Vurdering af hyppigheden af infektiøse-inflammatoriske patologier og antibiotikakurser i den ekstreme seksmånedersperiode;
- Røntgenbilleder af thorax med gentagne dynamikundersøgelser;
- vurdering af synkefunktion.
For patienter i den anden gruppe gælder følgende algoritme:
- fysisk eksamen;
- hjerte- og åndedrætsovervågning, polysomnografi til påvisning af pulmonal ventilationsmangel;
- pulsoximetri;
- Vurdering af hyppigheden af infektiøse-inflammatoriske processer og antibiotikakurser i den ekstreme periode på seks måneder;
- Undersøgelse af rygsøjlen, røntgen af rygsøjlen, vurdering af krumningsgraden.
Patienter i den tredje gruppe er indiceret til sådanne undersøgelser:
- fysisk eksamen;
- Respiratorisk funktionstest (omfatter spirometri, beregning af lungevolumen, vurdering af respiratorisk muskelfunktion);
- For at finde ud af hyppigheden af infektiøse-inflammatoriske patologier og antibiotikakurser i den ekstreme årlige periode.
Udøvelsen af differentialdiagnose kan være kompliceret af ligheden mellem SMN1 og SMN2 gener. For at undgå fejl anbefales det at bruge MLPA-metoden, som gør det muligt at detektere kopiantallet af exon 7 i SMN1-genet.
I de fleste tilfælde af spinal muskelatrofi er der en homozygot deletion af exon 7 og/eller 8 i SMN1-genet. Andre gener (ATP7A, DCTN1, UBA1, BSCL2, EXOSC3, GARS osv.) kan dog også være "syndere", hvilket man bør være opmærksom på, hvis SMN1-testen er negativ.
Biomaterialet til undersøgelsen kan være perifert blod eller føtalt blod, tørre blodpletkort. Diagnose er obligatorisk:
- i nærværelse af en forværret historie med spinal muskelatrofi;
- når der opdages mistænkelige symptomer, uanset arvelig historie.
Derudover anbefales forskning også til alle par, der er ansvarlige for at planlægge en graviditet.
Hvem skal kontakte?
Behandling Spinal muskelatrofi
Patienter med spinal muskelatrofi har brug for omfattende behandling, der omfatter:
- omsorg, hjælp, støtte;
- diæt mad;
- lægemiddelbehandling;
- ikke-medicinske rehabiliteringstiltag, herunder kinesioterapi og fysioterapi.
Et terapeutisk regime, der involverer en polymodal effekt på alle kropssystemer, ikke kun muskuloskeletale systemet, er standard.
Desværre er det umuligt radikalt at helbrede spinal muskelatrofi. Men det er ofte muligt at forbedre patientens livskvalitet gennem kompetent brug af aminosyrer og multivitaminkomplekser, neurotrofiske midler, calciumkanalblokkere, vasodilatorer, kardiotrofe og cytostatiske lægemidler, proteasehæmmere, steroide lægemidler, antioxidanter, immunglobuliner og immunsuppressiva, og snart. Det er eksperimentelt bevist, at behandling med stamceller, neurobeskyttende forbindelser og muskelstyrkende molekyler kan føre til uforudsigelige systemiske lidelser. Samtidig er positiv dynamik efter anvendelsen af en sådan behandling ikke blevet bevist hidtil.
Da problemet er forårsaget af en mangel på normalt SMN-protein, kan patienter forbedres ved at øge SMN-proteinniveauet med 25 % eller mere. Af denne grund forskes der aktivt i lægemidler, der kan aktivere produktionen af dette protein, herunder Gabapentin, Riluzol, Hydroxyurea, Albuterol, valproinsyre og natriumphenylbutyrat.
Moderne medicin tilbyder også kirurgisk behandling af spinal muskelatrofi. Det består af kirurgisk justering af rygsøjlen - korrektion af neuromuskulær krumning. Kirurger udfører multilevel fiksering af rygsøjlen ved hjælp af specielle konstruktioner. Korsbenet, bækkenet og hvirvlerne i den øvre thorax eller andre hvirvler bruges som støttepunkter. Operationen hjælper med at justere rygsøjlen, jævnt fordele belastningen på den, eliminere ubehag ved ændring af kroppens position, undgå negative virkninger på indre organer (inklusive lungerne).[12]
Medicin
I øjeblikket er der ingen ætiologisk behandling for spinal muskelatrofi: videnskabelig medicin fortsætter med at arbejde på denne opgave. Tidligere er det allerede lykkedes for forskere at isolere lægemidler, der kan øge produktionen af mRNA fra SMN2-genet. Men store internationale kliniske forsøg med personer med spinal muskelatrofi er endnu ikke blevet udført.
De fleste af de lægemidler, der indgår i standardbehandlingsregimet, har et generelt virkningsprincip med relativt lav evidens for effekt.
L-carnitin |
En naturligt forekommende aminosyre, en "slægtning" til B-gruppens vitaminer. Det produceres i kroppen, er til stede i leveren og tværstribede muskler, tilhører en række vitaminlignende stoffer. Deltager i metaboliske processer, understøtter CoA-aktivitet, bruges til at normalisere stofskiftet. Det har anabolsk, antithyroid, antihypoxisk evne, stimulerer lipidmetabolisme og vævsreparation, optimerer appetitten. L-Carnitin er ordineret i en mængde på omkring 1 tusind mg om dagen. Behandlingsforløbet kan vare op til 2 måneder. |
Coenzym Q10 (Ubiquinon) |
En coenzym benzoquinongruppe, der indeholder en række isoprenylgrupper. Disse er fedtopløselige coenzymer, hovedsageligt til stede i mitokondrier af eukaryote cellulære strukturer. Ubiquinon indgår i elektrontransportkæden, deltager i oxidativ phosphorylering. Den største tilstedeværelse af stoffet findes i energirige organer - især i leveren og hjertet. Coenzym Q10 har blandt andet antioxidante egenskaber, kan genoprette antioxidantkapaciteten af alfa-tocopherol. Normalt ordineret fra 30 til 90 mg af lægemidlet om dagen, et to-måneders kursus. |
Cerebrolysin |
Et nootropt lægemiddel med neurotrofiske egenskaber. Det bruges ofte i terapeutiske regimer til behandling af neurologiske patologier, herunder vaskulær demens, slagtilfælde. Den aktive fraktion inkluderer peptider med en begrænsende molekylvægt på 10 tusind dalton. Lægemidlet administreres som en intravenøs injektion på 1-2 ml. Behandlingsforløbet består af 10-15 injektioner. |
Actovegin |
Sammensætningen af lægemidlet er repræsenteret af lavmolekylære peptider og aminosyrederivater. Actovegin er et hæmoderivat: det isoleres ved dialyse med ultrafiltrering. Takket være brugen af lægemidlet øges absorptionen og udnyttelsen af ilt, energimetabolismen accelereres. Lægemidlet anvendes i form af intravenøse injektioner på 1-2 ml, kurset kræver 10-15 injektioner. |
Solcoseryl |
Det er et deproteiniseret hæmodialysat, der er i stand til at optimere præcellulær oxygen- og glucosetransport, øge intracellulær ATP-produktion, stimulere regenerative vævsreaktioner, aktivere fibroblastproliferation og kollagenproduktion i vaskulære vægge. Behandlingsforløbet består af 10-15 intramuskulære injektioner af lægemidlet (1-2 ml dagligt). |
Neuromultivit (vitamin B-kompleks) |
Multivitamin, aktivt brugt i mangel på vitaminer B-gruppe. Det er ofte i stand til at blive en kvalitetserstatning for et forløb med injektioner af vitaminpræparater. Aktiverer metaboliske processer i hjernen, fremmer genoprettelse af væv i nervesystemet, har en smertestillende effekt. Neuromultivit tage 1-2 tabletter dagligt, et forløb på 4 eller 8 uger. |
E-vitamin |
En velkendt antioxidant, fedtopløseligt vitamin. Det ordineres i forløb på 1-2 måneder i mængden af 10-20 IE dagligt. |
Valproat |
De har beroligende og afslappende aktivitet, demonstrerer antikonvulsiv evne, øger niveauet af GABA i CNS. Anvendes kun til behandling af børn over et år, 10 til 20 mg pr. Kg pr. Dag. |
Salbutamol |
En bronkodilatator, som tilhører gruppen af selektive beta2-adrenoreceptoragonister. Regelmæssig brug af lægemidlet forårsager øget produktion af mRNA og SMN-protein, hvilket positivt påvirker det kliniske billede af spinal muskelatrofi. Salbutamol anvendes med forsigtighed, 2-4 mg fire gange dagligt (maksimal mængde er 32 mg pr. Dag). |
Et af de nyeste lægemidler, der anvendes til spinal muskelatrofi, er Zolgensma® genotterapeutisk lægemiddel Zolgensma®, som sikrer aktiviteten og den korrekte funktion af transducerede motoriske nerveceller. Lægemidlet administreres i kombination med immunmodulerende lægemidler i henhold til en speciel protokol og administreres én gang intravenøst, baseret på en nominel dosis på 1,1 ͯ 1014 vg/kg (det samlede indgivelsesvolumen bestemmes afhængigt af patientens vægt).
Før behandling med Zolgensma påbegyndes, er det obligatorisk at bestemme niveauet af antistoffer mod AAV9 ved hjælp af en valideret diagnostisk metode, vurdere leverfunktionen (ALT, AST, total bilirubin), udføre generel klinisk blodundersøgelse og troponin I-test, bestemme kreatininniveauet. Hvis akutte og kroniske aktive infektionstilstande påvises, udsættes administrationen af lægemidlet indtil helbredelse eller afslutning af tilbagefaldsfasen af den infektionsproces.
Den hyppigste bivirkning af lægemidlet anses for at være leversvigt, som kan være dødelig.
Andre godkendte lægemidler, som din læge kan ordinere til spinal muskelatrofi:
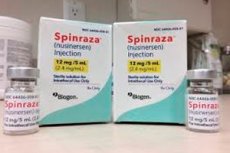
- Spinraza er et præparat af nusinersen-natrium, et antisense-oligonukleotid specielt designet til behandling af spinal amyotrofi. Det er beregnet til intratekal administration ved lumbalpunktur. Den anbefalede dosis er 12 mg. Behandlingsregimet bestemmes af den behandlende læge.
- Risdiplam er et lægemiddel, der modificerer splejsningen af mRNA-precursoren for det motoriske nervecelleoverlevelsesgen 2. Risdiplam tages oralt en gang dagligt. Doseringen bestemmes af lægen individuelt under hensyntagen til patientens alder og vægt. Brugen af lægemidlet til børn yngre end 2 måneder er kontraindiceret. Embryofetal toksicitet af dette lægemiddel er noteret, så reproduktionspotentielle patienter bør tage omhyggelige præventionsforanstaltninger både under og en periode efter behandlingen.
Fysioterapeutisk behandling af spinal muskelatrofi
Fysioterapi bruges som et af bindene i kompleks terapi og rehabilitering af patienter med spinal muskelatrofi. Hovedpunkterne i en sådan behandling er:
- brug af aflæsning ved hjælp af suspensionssystemer, aktiv-passiv træning, brug af perkutan elektrisk stimulering af rygmarven;
- åndedrætsøvelser og fysioterapi;
- halvtimes vertikaliseringssessioner;
- translingual elektrostimuleringsbehandlinger (20-minutters sessioner, kombineret med øvelser for at forbedre finmotorikken);
- manuelle teknikker;
- paraffinapplikationer på forskellige grupper af artikulationer;
- darsonval for at forbedre muskelydelsen.
Metoden til darsonvalisering er baseret på effekten på væv ved hjælp af en vekslende højfrekvent pulsstrøm med høj spænding og lav styrke. Efter et procedureforløb er der en stigning i muskelydelse, styrkelse af mikrocirkulationen, udvidelse af arterioler og kapillærer, eliminering af iskæmi, forbedring af ernæring og iltforsyning til muskler, hvilket har en positiv effekt på forløbet af regenerative og atrofiske processer.
Et af de mest betydelige problemer hos patienter med spinal amyotrofi er svækkelse af respiratoriske muskler, som ofte fører til respiratorisk dysfunktion og patientens død.
Ved spinal amyotrofi er hele skeletmuskulaturen, inklusive den der er ansvarlig for vejrtrækningen, underpræsterende. Svaghed og gradvis muskelatrofi påvirker kvaliteten af den respiratoriske handling negativt, fører til udvikling af komplikationer og øget respirationssvigt. Derfor er det nødvendigt at træffe foranstaltninger til at styrke musklerne, forebyggelse af luftvejskomplikationer og luftvejsinfektioner. En særlig rolle i dette spiller gymnastik med Ambu tasken, som udføres i forbindelse med fysioterapi, strækøvelser, massage. Brugen af Ambu taske giver dig mulighed for at "udvide" volumen af brystet og lungerne. Til børns aktiviteter er egnet taske med et volumen på mindst en og en halv liter, udstyret med en ventil til at frigive for højt tryk (for at forhindre barotraume).
Øvelser bør ikke udføres på en fuld mave. Kropsstilling - siddende, halvsiddende, liggende på siden eller ryggen (hvis der ikke er problemer med slim): det er optimalt at udføre procedurerne i forskellige stillinger hver gang. Det er vigtigt, at patientens ryg rettes op. Om nødvendigt bruges et korset. Før proceduren påbegyndes, skal du sikre dig, at luftvejen er fri for opspyt.
Massage til spinal muskelatrofi
Massage til behandling af spinal amyotrofi skal være let og blid. I områder med muskulær modstand gælder generelle effekter, herunder bankning, og i områder med bevaret innervation skal du bruge dybe strøg (på langs, på tværs), æltning.
Generelt, at praktisere forskellige typer massage, afhængigt af de individuelle karakteristika af sygdomsforløbet, patientens alder. Disse kan være:
- æltning for at stimulere dybtliggende muskler;
- gnider for at optimere blod- og lymfecirkulationen;
- pletbehandling af triggerpunkter;
- af den fiberstyrkende dunkende.
Det er vigtigt, at effekten spredes over hele problemområdet.
Kontraindikationer til massage for spinal muskelatrofi:
- akut betændelse, forhøjet kropstemperatur;
- blodsygdomme, blødningstendenser;
- purulente processer;
- infektionssygdomme, svampe dermatologiske sygdomme;
- Vaskulære aneurismer, trombangiitis, endarteritis, lymfadenitis;
- benigne og ondartede neoplasmer.
Forløbet af enhver massage til en patient med spinal muskelatrofi er ordineret strengt individuelt. Ukorrekt udførelse af proceduren, overdrevent grov og forkert påvirkning kan skade patientens tilstand.
Forebyggelse
Direkte og indirekte DNA-diagnostik og prænatal DNA-diagnose arbejdes der nu aktivt på. Dette reducerer markant sandsynligheden for, at en syg baby bliver født, hvilket især er vigtigt for par, der allerede har oplevet fødslen af børn med spinal muskelatrofi.
Forebyggende foranstaltninger repræsenterer en vigtig medicinsk tendens og er kategoriseret i primære, sekundære og tertiære foranstaltninger.
Primære foranstaltninger er rettet mod direkte at forhindre indflydelsen af en ugunstig faktor og forhindre udviklingen af sygdommen. Sådan forebyggelse består i at korrigere kost og daglige regimer, der fører en sund livsstil.
Sekundær forebyggelse består i eliminering af åbenlyse risikofaktorer og omfatter tidlig diagnose af patologier, etablering af overvågning i dynamik, rettet behandling.
Tertiær forebyggelse udføres i forhold til en syg person, der er frataget visse motoriske evner. I denne situation taler vi om medicin, psykologisk, social og arbejdsrehabilitering.
Ifølge oplysninger fra Verdenssundhedsorganisationen er mere end 2 % af babyer i verden født med en form for udviklingsforstyrrelse. Samtidig er 0,5-1% af sådanne lidelser af genetisk oprindelse. Forebyggelse af sådanne problemer er reduceret til medicinsk genetisk rådgivning og kvalitetsprænatal diagnose, som gør det muligt at minimere risikoen for at føde en baby med genetisk patologi.
En persons risiko for at få spinal muskelatrofi eller en anden genetisk sygdom afhænger af de gener, der er arvet fra hans mor og far. Tidlig identifikation af arvelige faktorer, beregning af individuelle risici for genetisk bestemt patologi kan kaldes en måde til målrettet forebyggelse.
Prænatale diagnostiske foranstaltninger omfatter direkte og indirekte forskningsmetoder. I første omgang identificeres kvinder, der kræver indirekte prænatal diagnose. Disse kan omfatte:
- gravide kvinder 35 år og ældre;
- som har haft 2 eller flere tidligere spontane aborter;
- som har børn med genetiske udviklingsdefekter;
- med en ugunstig arvelig historie;
- som har haft virusinfektioner eller strålingseksponering (inklusive i planlægningsfasen af graviditeten).
Til forebyggende formål anvendes sådanne metoder som ultralyd, hormonprøver (biokemisk screening). Nogle gange anvendes også invasive procedurer såsom chorionbiopsi, fostervandsprøve, placentocentese, cordocentesis. Pålidelig information om genetiske risici giver dig mulighed for at justere din livsstil og graviditet for at forhindre fødslen af et sygt barn.
Spinal muskelatrofivaccine
Selvfølgelig vil alle forældre til børn med spinal amyotrofi gerne helbrede dem fuldstændigt for sygdommen. Der er dog ingen vaccine, der kan udrydde problemet. Selvom forskningen i at optimere behandlingen er i gang.
Især i 2016 godkendte amerikanske videnskabsmænd det unikke lægemiddel Spinraza (nusinersen), som efterfølgende blev godkendt til brug i europæiske lande.
Specialister undersøger problemet med at behandle spinal muskelatrofi på disse måder:
- Reparation eller erstatning af det "forkerte" SMN1-gen;
- potensering af funktionen af det normale SMN2-gen;
- Beskyttelse af motoriske nerveceller påvirket på grund af SMN-proteinmangel;
- beskyttelse af muskler mod atrofiske ændringer for at forhindre eller genoprette tabt funktion på baggrund af patologisk udvikling.
Genterapi involverer målretning af det beskadigede gen ved hjælp af virale vektorer, der passerer gennem blod-hjerne-membranen og når det passende område i rygmarven. Derefter "inficerer" virussen den angrebne celle med en sund DNA-del, som om den "syede" genfejlen. Således er funktionen af motoriske nerveceller korrigeret.
En anden retning er terapi med små molekyler, hvis essens er at forbedre funktionen af SMN2-genet. Spædbørn med diagnosticeret spinal muskelatrofi har mindst én kopi af SMN2-genet. Denne retning er blevet aktivt undersøgt af amerikanske videnskabsmænd, og i øjeblikket gennemgår flere lægemidler, der har til formål at forbedre syntesen af et komplet protein fra SMN2-genet, kliniske forsøg.
En anden mulighed for mulig terapeutisk intervention er at udforske neurobeskyttelse for at reducere motorneurondød, øge deres adaptive kapacitet og forbedre funktionaliteten.
Den tredje retning involverer beskyttelse af musklen mod atrofiske processer. Da SMN-proteinmangel påvirker motoriske nerveceller og muskelfunktion negativt, bør målet med denne behandling være at beskytte musklerne mod atrofi, øge muskelmassen og genoprette muskelfunktionen. Denne type terapi vil ikke påvirke det genetiske apparat, men den kan bremse eller endda blokere forværringen af spinal muskelatrofi.
Screening for spinal muskelatrofi
Nyfødtscreening bruges i stigende grad i lægepraksis og spiller ofte en afgørende rolle. Opdagelse af spinal muskelatrofi så tidligt som muligt kan forbedre prognosen for det syge barn væsentligt. Screeningsdiagnose inkluderer følgende punkter, der er skitseret i tabellen:
En form for spinal muskelatrofi |
Symptomatologi |
Spinal muskelatrofi type I (barnet kan ikke sidde op, gennemsnitlig levetid - op til 2 år) |
Det viser sig fra fødslen til seks måneders alderen. Utilstrækkelig muskeltonus detekteres, gråden er svag, muskelsvaghed (inklusive tygge- og synkemuskler) øges. Der er problemer med hovedtilbageholdelse, babyen indtager en "frø" holdning, når den ligger ned. |
Spinal muskelatrofi type II (barnet er i stand til at sidde op, forventet levetid er normalt mere end 2 år, og mere end halvdelen af patienterne bliver 20-25 år gamle) |
Den debuterer fra 7 måneders alderen og op til halvandet års alderen. Synke-, åndedræts- og hosteproblemer bemærkes nogle gange. Permanente tegn inkluderer muskelspasmer, begrænset ledmobilitet, krumning af rygsøjlen, lavt blodtryk og muskelsvaghed. |
Spinal muskelatrofi type III (barnet kan sidde og bevæge sig, men ovenstående evner går gradvist tabt, forventet levetid er normal) |
Debuterer i en alder af halvandet år. Krumning af rygsøjlen og thorax, muskelatrofi af bækken og proksimale ben og øget ledmobilitet noteres. Synke er svært. |
Spinal muskelatrofi type IV |
Henviser til voksenformularen. Symptomatologi har meget til fælles med spinal muskelatrofi type III. Svagheden tiltager gradvist, rystelser og muskelfasciokulationer vises med debuten i 16-25 års alderen. |
Vejrudsigt
Ved Werdnig-Hoffmans syndrom er den gennemsnitlige levetid 1,5-2 år. Dødelig udgang skyldes i de fleste tilfælde øget respirationssvigt og udvikling af betændelse i lungerne. Med rettidig åndedrætsstøtte i form af kunstig ventilation er det muligt at øge babyens forventede levetid lidt. Der er et særligt behov for kontinuerlig palliativ behandling, hvilket også er påkrævet ved spinal amyotrofi type II. Patologier af den tredje og fjerde type er karakteriseret ved en mere gunstig prognose.
Enhver form for spinal muskelatrofi er en alvorlig sygdom. Alle familiemedlemmer til patienten kræver konstant psykologisk, informativ og social støtte. Det er vigtigt for patienten at sikre tilstrækkelig diagnose og professionel støtte fra speciallæger som børnelæge, neurolog, neurolog, lungelæge, kardiolog, ortopæd, fysioterapeut osv. På trods af manglende specifik terapi for sygdommen udføres symptomatisk behandling, særlig ernæring er ordineret (både parenteral og enteral), forskellige rehabiliteringsforanstaltninger, der bidrager til at bremse udviklingen af patologien og forhindre fremkomsten af komplikationer.
Mange patienter får tilkendt et handicap, og der udarbejdes en individuel rehabiliteringsordning.
Naturligt forekommende spinal muskelatrofi uden brug af særligt udstyr til støtte for vejrtrækning og fodring ender i omkring halvdelen af tilfældene med, at det syge barn dør før 2 års alderen (for det meste type I sygdom).