Nye publikationer
Nye resultater bidrager til en bedre forståelse af årsagerne til Rett syndrom
Sidst revideret: 02.07.2025

Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.

Rett syndrom er en sjælden neurologisk udviklingsforstyrrelse, som der i øjeblikket ikke findes nogen kur eller god behandling for. Det forårsager alvorlige fysiske og kognitive symptomer, hvoraf mange overlapper med autismespektrumforstyrrelser.
Rett syndrom skyldes mutationer i MECP2-genet, som er i høj grad udtrykt i hjernen og ser ud til at spille en vigtig rolle i at opretholde neuronernes sundhed. Genet er placeret på X-kromosomet, og syndromet rammer primært piger. For at udvikle behandlinger for Rett syndrom ønsker forskere at forstå MECP2 og dets funktioner i hjernen bedre.
Forskere, herunder medstifter af Whitehead Institute, Rudolf Jaenisch, har studeret MECP2 i årtier, men mange grundlæggende fakta om genet er forblevet ukendte. Proteinet, som genet koder for, MECP2, er involveret i genregulering; det binder sig til DNA og påvirker ekspressionsniveauerne af forskellige andre gener eller mængden af protein, de producerer.
Forskerne havde dog ikke en komplet liste over gener, der er påvirket af MECP2, og der var ingen konsensus om, hvordan MECP2 påvirker disse gener.
Tidlige studier af MECP2 antydede, at det var en repressor, der reducerede ekspressionen af dets målgener, men forskning foretaget af Jaenisch og andre havde tidligere vist, at MECP2 også fungerer som en aktivator, der øger ekspressionen af dets mål – og at det muligvis er en aktivator i første omgang. Også ukendt var MECP2's virkningsmekanisme, eller hvad proteinet præcist gør for at forårsage ændringer i genekspression.
Teknologiske begrænsninger har forhindret forskere i at få klarhed over disse spørgsmål. Men Yanish, hans laboratoriepostdoc Yi Liu, og Yanishs tidligere laboratoriemedlem Anthony Flamier, nu adjunkt ved CHU Sainte-Justine forskningscenter på Université de Montréal, har brugt banebrydende teknikker til at besvare disse resterende spørgsmål om MECP2 og få ny indsigt i dets rolle i hjernens sundhed og sygdom.
Deres resultater blev offentliggjort i tidsskriftet Neuron, og forskerne oprettede også et online arkiv med deres MECP2-data, MECP2-NeuroAtlas-portalen, som en ressource for andre forskere.
"Jeg tror, at denne artikel fundamentalt vil ændre folks forståelse af, hvordan MECP2 forårsager Rett syndrom. Vi har en helt ny forståelse af mekanismen, og den kan give nye muligheder for at udvikle behandlinger for sygdommen," siger Janisch, der også er professor i biologi ved MIT.
Dybere forståelse af MECP2 i hjernen
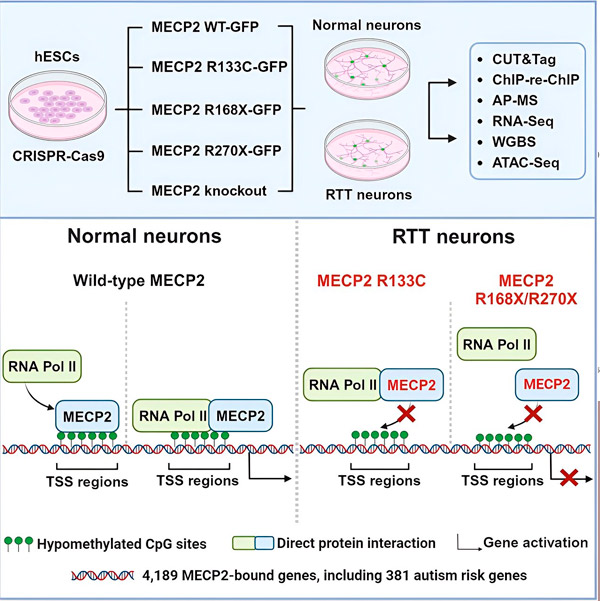
Forskerne skabte først et detaljeret kort over, hvor MECP2 binder sig i humane neuronale gensekvenser, enten inden for gener eller i regulatoriske regioner af DNA i nærheden af dem. De brugte en metode kaldet CUT&Tag, som kan lokalisere proteininteraktioner med DNA med høj præcision.
Forskerne fandt mere end 4.000 gener forbundet med MECP2. De gentog deres kortlægning i neuroner med almindelige MECP2-mutationer forbundet med Rett syndrom for at bestemme, hvor MECP2 er udtømt i sygdomstilstanden.
Da Liu og Flamier vidste, hvilke gener MECP2 binder sig til, kunne de begynde at skabe forbindelser mellem MECP2's mål og hjernens sundhed. De fandt ud af, at mange af dets mål er involveret i udviklingen og funktionen af neuronale axoner og synapser.
De sammenlignede også deres liste over MECP2-mål med Simons Foundation Autism Research Initiative (SFARI) database over autisme-associerede gener og fandt, at 381 gener i denne database er MECP2-mål.
Kilde: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Disse fund kan bidrage til at afklare de mekanismer, der ligger til grund for autismesymptomer ved Rett syndrom, og give et godt udgangspunkt for at undersøge MECP2's mulige rolle i autisme.
"Vi har skabt det første integrerede kort over MECP2-epigenomet i sundhed og sygdom, og dette kort kan vejlede fremtidig forskning," siger Liu. "At vide, hvilke gener der er mål for MECP2, og hvilke gener der er direkte forstyrret i sygdommen, giver et solidt fundament for at forstå Rett syndrom og stille spørgsmål om genregulering i neuroner."
Forskerne undersøgte også, om MECP2 øgede eller mindskede ekspressionen af sine målgener. I overensstemmelse med historien om, at MECP2 af nogle er blevet identificeret som en aktivator og af andre som en repressor, fandt Liu og Flamier eksempler, hvor MECP2 spillede begge roller.
Men selvom MECP2 oftere opfattes som en repressor, fandt Liu og Flamier, at det primært er en aktivator – hvilket bekræfter tidligere fund foretaget af Jaenisch og Liu. Et nyt eksperiment viste, at MECP2 aktiverer mindst 80% af sine mål, og et andet viste, at det aktiverer op til 88% af sine mål.
Kortet over målgener, som forskerne skabte, gav yderligere indsigt i MECP2's rolle som aktivator. De fandt ud af, at for gener, som MECP2 aktiverer, binder det typisk til en region af DNA opstrøms for genet kaldet transkriptionsstartstedet.
Dette er stedet, hvor cellulært maskineri initierer processen med at transkribere et gen til RNA, hvorefter RNA'et translateres til et funktionelt protein, som er produktet af genekspression. Tilstedeværelsen af MECP2 på transkriptionsstartstedet, hvor genekspression begynder, er i overensstemmelse med dets rolle som genaktivator.
Forskerne satte sig derefter for at bestemme, hvilken rolle MECP2 spiller i genaktivering. De undersøgte, hvilke molekyler MECP2 binder til på dette sted, udover DNA, og fandt ud af, at MECP2 interagerer direkte med et proteinkompleks kaldet RNA polymerase II (RNA Pol II). RNA Pol II er en vigtig cellulær maskine, der transkriberer DNA til RNA. RNA Pol II kan ikke finde gener på egen hånd, så det kræver en række kofaktorer, eller proteinkollaboratører, for at hjælpe det med at udføre sit arbejde.
Forskerne foreslår, at MECP2 fungerer som en sådan cofaktor, der hjælper RNA Pol II med at initiere transkription i gener, hvor MECP2 binder. Strukturel analyse af MECP2 har identificeret dele af molekylet, der binder til RNA Pol II, og andre eksperimenter har bekræftet, at tab af MECP2 reducerer tilstedeværelsen af RNA Pol II på passende transkriptionsstartsteder, såvel som ekspressionsniveauerne af målgener.
Dette tyder på, at Rett syndrom kan være forårsaget af nedsat transkription af gener, som MECP2 er målrettet mod, på grund af MECP2-mutationer, der forhindrer det i at binde sig til RNA Pol II eller binde sig til DNA. I overensstemmelse med denne idé er de mest almindelige MECP2-mutationer forbundet med sygdom trunkeringer: mutationer, hvor en del af proteinet mangler, hvilket kan ændre interaktionen mellem MECP2 og RNA Pol II.
Forskerne håber, at deres resultater ikke blot vil ændre vores forståelse af MECP2, men at en dybere og bredere forståelse af, hvordan MECP2 påvirker hjernens udvikling og funktion, kan føre til nye indsigter, der vil hjælpe mennesker med Rett syndrom og relaterede lidelser, herunder autisme.
"Dette projekt er et godt eksempel på Janisch-laboratoriets samarbejdsorienterede natur," siger Flamier. "Rudolf og jeg havde et specifikt problem relateret til Rett syndrom, og jeg havde erfaring med CUT&Tag-teknologien, som kunne løse problemet. Gennem diskussion indså vi, at vi kunne kombinere vores indsats, og nu har vi et stort lager af information om MECP2 og dets forbindelse til sygdom."